Nature Ecology & Evolution ( IF 13.9 ) Pub Date : 2024-11-20 , DOI: 10.1038/s41559-024-02571-w Yoav Mathov, Malka Nissim-Rafinia, Chen Leibson, Nir Galun, Tomas Marques-Bonet, Arye Kandel, Meir Liebergal, Eran Meshorer, Liran Carmel
|
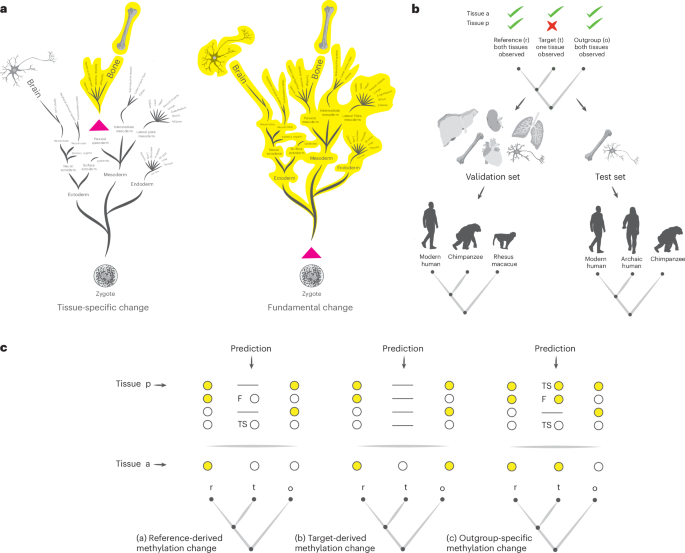
Genome-wide premortem DNA methylation patterns can be computationally reconstructed from high-coverage DNA sequences of ancient samples. Because DNA methylation is more conserved across species than across tissues, and ancient DNA is typically extracted from bones and teeth, previous works utilizing ancient DNA methylation maps focused on studying evolutionary changes in the skeletal system. Here we suggest that DNA methylation patterns in one tissue may, under certain conditions, be informative on DNA methylation patterns in other tissues of the same individual. Using the fact that tissue-specific DNA methylation builds up during embryonic development, we identified the conditions that allow for such cross-tissue inference and devised an algorithm that carries it out. We trained the algorithm on methylation data from extant species and reached high precisions of up to 0.92 for validation datasets. We then used the algorithm on archaic humans, and identified more than 1,850 positions for which we were able to observe differential DNA methylation in prefrontal cortex neurons. These positions are linked to hundreds of genes, many of which are involved in neural functions such as structural and developmental processes. Six positions are located in the neuroblastoma breaking point family (NBPF) gene family, which probably played a role in human brain evolution. The algorithm we present here allows for the examination of epigenetic changes in tissues and cell types that are absent from the palaeontological record, and therefore provides new ways to study the evolutionary impacts of epigenetic changes.
中文翻译:
推断古代标本非骨骼组织中的 DNA 甲基化
全基因组死前 DNA 甲基化模式可以从古代样本的高覆盖率 DNA 序列进行计算重建。由于 DNA 甲基化在物种之间比跨组织更保守,并且古代 DNA 通常从骨骼和牙齿中提取,因此以前利用古代 DNA 甲基化图谱的工作侧重于研究骨骼系统的进化变化。在这里,我们认为,在某些条件下,一个组织中的 DNA 甲基化模式可能为同一个体的其他组织中的 DNA 甲基化模式提供信息。利用组织特异性 DNA 甲基化在胚胎发育过程中积累的事实,我们确定了允许这种跨组织推断的条件,并设计了一种执行它的算法。我们在现存物种的甲基化数据上训练了算法,验证数据集的精度高达 0.92。然后,我们在古人类身上使用了该算法,并确定了 1,850 多个位置,我们能够在前额叶皮层神经元中观察到不同的 DNA 甲基化。这些位置与数百个基因有关,其中许多基因与结构和发育过程等神经功能有关。神经母细胞瘤断裂点家族 (NBPF) 基因家族中有 6 个位置,可能在人脑进化中发挥作用。我们在这里介绍的算法允许检查古生物学记录中不存在的组织和细胞类型的表观遗传变化,因此为研究表观遗传变化的进化影响提供了新的方法。






























 京公网安备 11010802027423号
京公网安备 11010802027423号