当前位置:
X-MOL 学术
›
Chem. Mater.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Exploring Molecules with Low Viscosity: Using Physics-Based Simulations and De Novo Design by Applying Reinforcement Learning
Chemistry of Materials ( IF 7.2 ) Pub Date : 2024-11-19 , DOI: 10.1021/acs.chemmater.4c02929 Nobuyuki N. Matsuzawa, Hiroyuki Maeshima, Keisuke Hayashi, Tatsuhito Ando, Mohammad Atif Faiz Afzal, Kyle Marshall, Benjamin J. Coscia, Andrea R. Browning, Alexander Goldberg, Mathew D. Halls, Karl Leswing, Mayank Misra, Farhad Ramezanghorbani, Tsuguo Morisato
Chemistry of Materials ( IF 7.2 ) Pub Date : 2024-11-19 , DOI: 10.1021/acs.chemmater.4c02929 Nobuyuki N. Matsuzawa, Hiroyuki Maeshima, Keisuke Hayashi, Tatsuhito Ando, Mohammad Atif Faiz Afzal, Kyle Marshall, Benjamin J. Coscia, Andrea R. Browning, Alexander Goldberg, Mathew D. Halls, Karl Leswing, Mayank Misra, Farhad Ramezanghorbani, Tsuguo Morisato
|
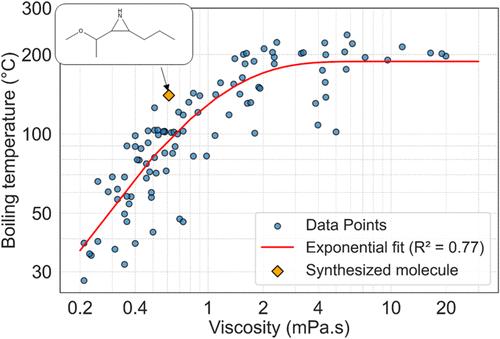
Molecules with viscosities lower than those of conventional organic solvents are highly sought after for applications in electrochemical devices such as batteries and capacitors. These molecules improve the electrical resistance of devices, enhancing their efficiency, especially at low temperatures. To identify new molecules with low viscosities, we conducted extensive molecular dynamics (MD) simulations on 10,000 molecules selected from the GDB-17 chemical structure database, specifically choosing molecules with fewer than 12 heavy atoms. Additionally, we performed density functional theory (DFT) calculations to determine the energies of the highest occupied molecular orbitals (HOMO) of these molecules as a surrogate for the oxidation potential. We used the data on viscosity and HOMO levels as training sets to develop machine-learning models that predict these properties. Using these models, we carried out molecular de novo design using the REINVENT method, a reinforcement-learning approach utilizing SMILES strings. This method aimed to identify molecules that minimize viscosity while maintaining sufficiently low HOMO levels for stability. The approach successfully identified new chemical structures with viscosities below 2 mPa·s and suitably low HOMO energies. We synthesized a novel compound from the top candidates and validated our predictions experimentally. The experimental results closely matched our predictions, demonstrating that combining physics-based simulations with reinforcement learning is an effective strategy for designing novel molecules with targeted properties.
中文翻译:

探索低粘度分子:通过应用强化学习使用基于物理的仿真和从头设计
粘度低于传统有机溶剂的分子在电池和电容器等电化学设备中的应用备受追捧。这些分子提高了器件的电阻,提高了它们的效率,尤其是在低温下。为了鉴定低粘度的新分子,我们对从 GDB-17 化学结构数据库中选择的 10,000 个分子进行了广泛的分子动力学 (MD) 模拟,特别是选择了重原子少于 12 个的分子。此外,我们进行了密度泛函理论 (DFT) 计算,以确定这些分子的最高占据分子轨道 (HOMO) 的能量,作为氧化电位的替代物。我们使用粘度和 HOMO 水平数据作为训练集来开发预测这些特性的机器学习模型。使用这些模型,我们使用 REINVENT 方法进行了分子从头设计,这是一种利用 SMILES 字符串的强化学习方法。该方法旨在鉴定在保持足够低的 HOMO 水平以实现稳定性的同时最大限度地降低粘度的分子。该方法成功鉴定了粘度低于 2 mPa·s 且具有适当低 HOMO 能量的新化学结构。我们从顶级候选化合物中合成了一种新型化合物,并通过实验验证了我们的预测。实验结果与我们的预测非常吻合,表明将基于物理的模拟与强化学习相结合是设计具有目标特性的新型分子的有效策略。
更新日期:2024-11-19
中文翻译:
探索低粘度分子:通过应用强化学习使用基于物理的仿真和从头设计
粘度低于传统有机溶剂的分子在电池和电容器等电化学设备中的应用备受追捧。这些分子提高了器件的电阻,提高了它们的效率,尤其是在低温下。为了鉴定低粘度的新分子,我们对从 GDB-17 化学结构数据库中选择的 10,000 个分子进行了广泛的分子动力学 (MD) 模拟,特别是选择了重原子少于 12 个的分子。此外,我们进行了密度泛函理论 (DFT) 计算,以确定这些分子的最高占据分子轨道 (HOMO) 的能量,作为氧化电位的替代物。我们使用粘度和 HOMO 水平数据作为训练集来开发预测这些特性的机器学习模型。使用这些模型,我们使用 REINVENT 方法进行了分子从头设计,这是一种利用 SMILES 字符串的强化学习方法。该方法旨在鉴定在保持足够低的 HOMO 水平以实现稳定性的同时最大限度地降低粘度的分子。该方法成功鉴定了粘度低于 2 mPa·s 且具有适当低 HOMO 能量的新化学结构。我们从顶级候选化合物中合成了一种新型化合物,并通过实验验证了我们的预测。实验结果与我们的预测非常吻合,表明将基于物理的模拟与强化学习相结合是设计具有目标特性的新型分子的有效策略。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号