当前位置:
X-MOL 学术
›
J. Org. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
From Established to Emerging: Evolution of Cross-Coupling Reactions
The Journal of Organic Chemistry ( IF 3.3 ) Pub Date : 2024-11-15 , DOI: 10.1021/acs.joc.4c02573 Mark R. Biscoe, Josep Cornella, Dipannita Kalyani, Sharon Neufeldt
The Journal of Organic Chemistry ( IF 3.3 ) Pub Date : 2024-11-15 , DOI: 10.1021/acs.joc.4c02573 Mark R. Biscoe, Josep Cornella, Dipannita Kalyani, Sharon Neufeldt
|
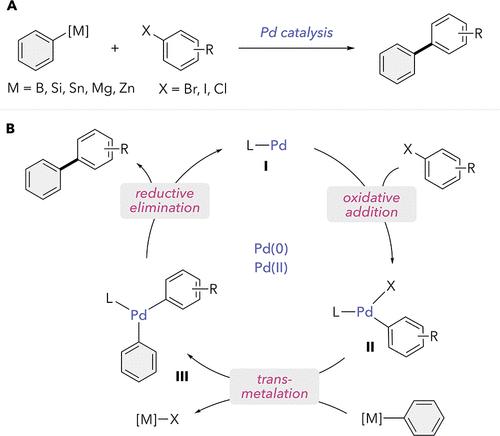
This article is part of the Next-Generation Cross-Coupling Chemistry special issue. The construction of bonds between C atoms and biologically relevant elements such as C, O, N, S, and P (among others) is a fundamental goal of synthetic organic chemists. Despite early advances in C–C bond forming reactions (e.g., aldol chemistry, reactions of Grignard reagents), in the 1960s a key challenge remained: the efficient construction of C–C bonds between two C(sp2) atoms (Figure 1A). By the late 1970s, the work of multiple chemists converged on a solution involving the coupling of alkenes or aryl nucleophiles derived from main group organometallic species [C(sp2)–M; M = B, Sn, Zn, or Mg] with aryl electrophiles [C(sp2)–X; X = I, Br, Cl, OTf] in the presence of a Ni or Pd catalyst. (1−8) With these discoveries, a new paradigm for the formation of chemical bonds emerged. The importance and impact of the C–C cross-coupling reaction catapulted it to a venerable place in the field of synthesis, together with amide bond formation and nucleophilic substitution. (9) In 2010, the Nobel Prize in Chemistry was awarded to contributors who spearheaded this revolution, namely, Akira Suzuki, Ei-ichi Negishi, and Richard F. Heck. (10) Figure 1. (A) Pd-catalyzed cross-coupling reaction. (B) Canonical mechanism of cross-couplings involving organometallic nucleophiles. Although the origin of the term “cross-coupling” is commonly associated with the development of the Suzuki–Miyaura, Stille, Kumada, and Negishi reactions (named after their inventors), earlier examples existed from Glaser, (11) Ullman, (12) or Kharasch (13) involving reactions between organometallic reagents and aryl halides catalyzed by transition metal salts. However, the low selectivity and yields and the lack of mechanistic understanding limited the widespread adoption of such strategies. On the other hand, Pd-catalyzed cross-coupling reactions offered high yields, broadened substrate scope, and the possibility to isolate and study stable organometallic intermediates. Over the years, numerous mechanistic investigations of transition-metal-catalyzed cross-coupling reactions have appeared in the literature. Based on these studies, the community reached an agreement that the catalytic manifold for cross-couplings involving organometallic nucleophiles includes three main organometallic steps: oxidative addition, transmetalation, and reductive elimination (Figure 1B). Although each mechanistic step has its own intricacies and complications, this general picture has served organic chemists as a guiding principle by which to understand and predict reactivity, selectivity, and other features to advance the field of cross-coupling. From the continuous evolution of cross-coupling strategies, a dizzying diversity of mechanisms, reagent classes, and product structures characterizes modern cross-couplings. As these reactions stray from the traditional characteristics of canonical cross-couplings, the term “cross-coupling” has evolved to represent a synthetic philosophy rather than a mechanistic or reactant-bound definition. (14) In a general sense, cross-coupling reactions can be simply illustrated as merging two (or more) building blocks together to construct new bond(s). The reactions are mediated by a third party, an entity that is traditionally a transition metal catalyst, but is occasionally very different (e.g., photons, (15) a main group element (16)). Reactions that cannot be easily placed into a different well-defined category can often be good candidates for inclusion in the broader “cross-coupling” framework. Inspired by the creative reports in this Special Issue, here we attempt to identify some of the research themes that have characterized the field of cross-coupling. Along the left side of the timeline in Figure 2, points are marked to indicate seminal publication(s) for each theme. (1,3,10−12,17−33) In more ambiguous cases, landmark publications were chosen based on the resemblance between the products and those of canonical cross-couplings. For example, Périchon’s synthesis of biaryls in 1993 was selected to represent the beginning of the era of electrochemical cross-coupling. (24) For many themes, multiple years─or even decades─passed before a relative flurry of activity on the theme began to appear in the literature. The points along the right side of the timeline represent some publications that contributed to the inflection period in popularity of a theme. (2,4−8,13,34−48) For brevity, we selected a minimal number of examples, but this list of key contributions is certainly incomplete and numerous other papers represent important strides within the research areas. Many of the reports in this Special Issue fall into one or more of the research themes illustrated in the timeline. Other articles, such as those by Li and Sun (DOI: 10.1021/acs.joc.3c02417), Lv and Li (DOI: 10.1021/acs.joc.3c00744), Shenvi (DOI: 10.1021/acs.joc.4c00260), and co-workers, branch out even further and highlight the idea of cross-coupling as a synthetic philosophy. Figure 2. Timeline of selected research themes in the area of cross-coupling (XC): seminal work (left side) and approximate inflection periods (right side). While cross-coupling reactions were initially largely dominated by the use of aryl iodides and bromides, the desire to broaden the scope to include more challenging electrophiles, such as Ar–Cl and phenolic derivatives like Ar–OTs and Ar–OMs, sparked innovations in ligand design and in Ni-catalyzed cross-couplings. Since the original reports from Wenkert demonstrating Ni-catalyzed Kumada cross-couplings with Ar–OMe and Ar–OCOR, (20) a diversity of methods have been reported for employing challenging electrophiles, typically mediated by highly nucleophilic Ni catalysts (44) or by Pd supported by bulky ligands. In this issue, reports by Johnson and Stradiotto (DOI: 10.1021/acs.joc.3c01584) and by Szostak (DOI: 10.1021/acs.joc.4c00103) represent examples of cross-couplings that involve activation of strong bonds. As more classes of electrophiles have become viable coupling partners for cross-coupling reactions, issues of chemo- and site-selectivity needed to be addressed, (28,45) as exemplified in a report by So et al. (DOI: 10.1021/acs.joc.3c02345). Fundamental mechanistic studies on single organometallic catalytic steps, such as an article in this issue from Denmark et al. (DOI: 10.1021/acs.joc.3c02629), continue to fuel advances in cross-couplings. Concurrent with establishment of the canonical mechanism in Figure 1B, Heck and Sonogashira cross-couplings emerged, illustrating that deviations from the standard pathway are also possible. (1,2,19,38) These mechanistic variations spurred the creativity of synthetic chemists, resulting in several modifications of the classical cross-coupling paradigm. For example, replacing prefunctionalized nucleophiles with simple C–H nucleophiles has led to a major revolution in the field of organic synthesis. (22,40) A contribution in this issue by Hruszkewycz, Leitch, and co-workers exploits this theme for the direct alkenylation of heterocycles (DOI: 10.1021/acs.joc.3c02311). Even more drastic deviations from the canonical mechanism have arisen from the recognition that cross-coupling can be achieved through single-electron transfer (SET) chemistry. Such processes perhaps represent one of the most active research areas in cross-coupling currently, and can be achieved through chemical, photochemical, or electrochemical methods. In this Special Issue, a majority of manuscripts fit into one of these themes characterized by radical species, including Perspectives by Bahamonde (DOI: 10.1021/acs.joc.3c02353), Gesmundo (DOI: 10.1021/acs.joc.3c02351), and co-workers. Though early studies of metal-catalyzed cross-coupling reactions focused on the formation of C(sp2)–C(sp2) bonds, successful incorporation of C(sp3) nucleophiles and electrophiles into cross-coupling methods has more recently enabled researchers to develop a broad array of strategies to elaborate three-dimensional organic molecules via C(sp3)–C(sp2) or C(sp3)–C(sp3) bond-forming processes. (17,25,36,43) To circumvent the inherent challenge of alkyl transmetalation and oxidative addition, the development of new approaches to efficiently transfer alkyl groups to transition metal catalysts is required. Because alkyl nucleophiles and alkyl electrophiles can each be incorporated into the catalytic cycle via one-electron or two-electron mechanisms, a wide array of cross-coupling approaches can be envisioned using a variety of transition metal catalysts and cross-coupling mediators. Modern strategies capitalizing on SET mechanisms have been particularly impactful in facilitating the development of new C(sp3)–C(sp2) and C(sp3)–C(sp3) bond-forming reactions. Collectively, these new approaches are well represented in the current issue where numerous cross-coupling studies feature reactions involving at least one alkyl partner. In this Special Issue, Denmark (DOI: 10.1021/acs.joc.4c00089) describes a Pd-catalyzed C(sp3)–C(sp2) bond-forming Suzuki–Miyaura reaction involving primary alkylboronic esters that undergo transmetalation via a two-electron mechanism. Noël and Watson each report studies involving the use of Ni-catalyzed alkyl deamination to access alkyl radicals for use in cross-coupling reactions. Noël (DOI: 10.1021/acs.joc.3c00859) incorporates alkyl groups into a Ni-catalyzed cross-electrophile reaction via a SET deamination pathway enabled by electrocatalysis. In contrast, Watson (DOI: 10.1021/acs.joc.3c00665) employs a Ni-catalyzed approach that selectively couples alkylzinc nucleophiles with alkyl partners generated by deamination. Both Waldvogel and Rousseaux describe new Ni-catalyzed cross-coupling reactions in which generation of alkyl radicals is achieved through the use of redox-active esters. Waldvogel (DOI: 10.1021/acs.joc.4c00428) combines Ni catalysis and electrocatalysis to generate primary alkyl radicals from redox-active esters for use in the synthesis of sitagliptin. Rousseaux (DOI: 10.1021/acs.joc.3c02354) employs reductive Ni-catalyzed cross-coupling of redox-active esters and aryl iodides for the preparation of α-aryl nitriles. The works of Xiao, Shen, and Roberts all engage alkyl radicals in cross-coupling reactions through the merger of transition metal catalysis and visible light catalysis. In the work of Xiao (DOI: 10.1021/acs.joc.3c02348), tertiary alkyl radicals are generated photocatalytically from alkylgermanium nucleophiles and incorporated into subsequent Ni-catalyzed acylation reactions. Shen (DOI: 10.1021/acs.joc.3c02293) merges Ni-catalysis and photocatalysis in a stereoconvergent reductive coupling that enables the formation of highly enantioenriched benzylic alcohols. Roberts (DOI: 10.1021/acs.joc.3c00872), in a conceptually novel approach to the formation of alkyl radicals, merges alkyl decarboxylation and alkyl desulfonylation in a photocatalyzed Cu-mediated cross-coupling approach toward benzylic trifluoromethylation. Finally, Giri (DOI: 10.1021/acs.joc.3c02548) describes a process for the dibenzylation of styrenes that proceeds via the iron-promoted formation of benzylic radicals. SET pathways are also useful for next-generation cross-couplings not involving alkyl coupling partners. For example, in this issue Li, Sun, and Ding (DOI: 10.1021/acs.joc.4c00021) employ electrochemical cross-coupling to access biaryl products. Kakiuchi (DOI: 10.1021/acs.joc.3c02601) describes the use of electrochemistry in combination with palladium catalysis to achieve C–H iodination. Alongside the evolution of diverse paradigms, new technologies such as High-Throughput Experimentation (HTE) emerged to best leverage the potential of cross-couplings. (29,49) HTE has become a mainstay in process and medicinal chemistry campaigns, enabling rapid reaction optimization and exploration of chemical space through parallel library synthesis. HTE can be leveraged for multiparameter optimization of reaction conditions for one combination of coupling partners, as illustrated in a report in this issue by Bock and Denmark (DOI: 10.1021/acs.joc.4c00089). Alternatively, HTE can be used for the simultaneous screen of reaction conditions for a diverse range of coupling partners, as discussed by Gesmundo and co-workers at AbbVie (DOI: 10.1021/acs.joc.3c02351) and by Leitch (DOI: 10.1021/acs.joc.3c02311) in partnership with researchers at GSK. The latter screens often reveal a set of reaction conditions that collectively enable access to a broader chemical space than any singular method. While HTE has been leveraged for exploring thermal reactions for over a decade, its adoption for photochemical and electrochemical transformations have only surfaced in recent years. As illustrated in the Perspective by Gesmundo et al. (DOI: 10.1021/acs.joc.3c02351), the widespread adoption of HTE for photochemical transformations required systematic exploration of both equipment and reaction conditions to identify processes that are user-friendly, robust, and lead to high success rates with a broad range of coupling partners. With the advent of HTE equipment for electrochemistry, a similar systematic approach will likely be necessary for the seamless adoption of high-throughput electrochemical transformations across industry and academia. While HTE has enabled the expedited exploration of chemical space, screening remains time and resource intensive. Hence a forward-looking approach entails the use of HTE data to build and apply predictive ML models for synthetic transformations. (32) The earliest paradigms of cross-coupling transformations have inspired over a century of innovations spanning diverse directions. Even as modern chemists continue to advance the more established cross-coupling research themes, these progressively branch into new directions. In parallel, the past few decades have witnessed innovations in technologies such as HTE to accelerate the optimization and discovery of the next generation of cross-couplings across academia and industry, often in partnership. We anticipate that future innovations, including data science and discoveries of new mechanistic manifolds, will continue to expand the diversity of cross-coupling chemistry. This article references 49 other publications. For a recent pertinent review, see: This article has not yet been cited by other publications.
中文翻译:

从成熟到新兴:交叉偶联反应的演变
本文是 Next-Generation Cross-Coupling Chemistry 特刊的一部分。在 C 原子和生物相关元素(如 C、O、N、S 和 P(等)之间构建键是合成有机化学家的基本目标。尽管 C-C 键形成反应(例如,羟醛化学、格氏试剂的反应)取得了早期进展,但在 1960 年代,一个关键挑战仍然存在:在两个 C(sp2) 原子之间有效构建 C-C 键(图 1A)。到 1970 年代后期,多位化学家的工作集中在一种解决方案上,该解决方案涉及来自主要族有机金属物种 [C(sp2)–M;M = B、Sn、Zn 或 Mg] 与芳基亲电试剂 [C(sp2)–X;X = I、Br、Cl、OTf] 在 Ni 或 Pd 催化剂存在下。(1−8) 随着这些发现,出现了形成化学键的新范式。C-C 交叉偶联反应的重要性和影响使其与酰胺键形成和亲核取代一起在合成领域一跃成为受人尊敬的地位。(9) 2010 年,诺贝尔化学奖颁发给了引领这场革命的贡献者,即铃木明、根岸荣一和理查德 F. 赫克。(10) 图 1.(A) Pd 催化的交叉偶联反应。(B) 涉及有机金属亲核试剂的交叉偶联的经典机制。尽管术语“交叉偶联”的起源通常与 Suzuki-Miyaura、Stille、Kumada 和 Negishi 反应(以其发明者命名)的发展有关,但早期的例子存在于 Glaser、(11)、Ullman、(12) 或 Kharasch (13) 涉及有机金属试剂和芳基卤化物之间的反应由过渡金属盐催化。 然而,低选择性和产量以及缺乏对机械的理解限制了此类策略的广泛采用。另一方面,Pd 催化的交叉偶联反应提供了高产率、拓宽了底物范围以及分离和研究稳定有机金属中间体的可能性。多年来,文献中出现了许多过渡金属催化交叉偶联反应的机理研究。基于这些研究,社区达成了一项协议,即涉及有机金属亲核试剂的交叉偶联的催化歧管包括三个主要的有机金属步骤:氧化加成、金属转移和还原消除(图 1B)。尽管每个机理步骤都有其自身的复杂性和复杂性,但这一总体情况为有机化学家提供了理解和预测反应性、选择性和其他特征的指导原则,以推进交叉偶联领域。从交叉偶联策略的不断发展来看,令人眼花缭乱的机制、试剂类别和产品结构的多样性是现代交叉偶联的特点。由于这些反应偏离了规范交叉偶联的传统特征,因此术语“交叉偶联”已经演变为代表一种合成哲学,而不是机理或反应物结合的定义。(14) 从一般意义上讲,交叉偶联反应可以简单地描述为将两个(或多个)构建单元合并在一起以构建新的键。这些反应由第三方介导,第三方是传统上是过渡金属催化剂的实体,但偶尔会非常不同(例如,光子,(15) 一个主要的族元素 (16))。 不能轻易归入定义明确的不同类别的反应通常可以很好地纳入更广泛的“交叉偶联”框架。受本期特刊中创意报告的启发,我们在这里尝试确定交叉耦合领域的一些研究主题。在图 2 中,沿着时间线的左侧,标记了一些点以指示每个主题的开创性出版物。(1,3,10−12,17−33)在更模糊的情况下,根据产物与规范交叉偶联的产物之间的相似性来选择具有里程碑意义的出版物。例如,Périchon 在 1993 年合成的联芳基被选为代表电化学交叉偶联时代的开始。(24) 对于许多主题来说,多年甚至几十年后,关于该主题的相对一连串活动才开始出现在文献中。时间轴右侧的点表示一些出版物,这些出版物导致了主题流行度的拐点期。(2,4−8,13,34−48)为简洁起见,我们选择了最少数量的例子,但这份关键贡献清单肯定是不完整的,许多其他论文代表了研究领域内的重要进步。本期特刊中的许多报告都属于时间轴中说明的一个或多个研究主题。其他文章,如 Li 和 Sun (DOI: 10.1021/acs.joc.3c02417)、Lv 和 Li (DOI: 10.1021/acs.joc.3c00744)、Shenvi (DOI: 10.1021/acs.joc.4c00260) 及其同事的文章,进一步扩展并强调了交叉耦合作为一种综合哲学的想法。图 2.交叉偶联 (XC) 领域选定研究主题的时间表:开创性工作(左侧)和近似拐点(右侧)。 虽然交叉偶联反应最初主要以芳基碘化物和溴化物的使用为主,但希望扩大范围以包括更具挑战性的亲电试剂(如 Ar-Cl)和酚类衍生物(如 Ar-OT 和 Ar-OM),这激发了配体设计和 Ni 催化交叉偶联的创新。自从 Wenkert 的原始报告证明了 Ni 催化的 Kumada 与 Ar-OMe 和 Ar-OCOR 的交叉偶联 (20) 以来,已经报道了多种使用具有挑战性的亲电试剂的方法,通常由高亲核性 Ni 催化剂 (44) 或由大体积配体支持的 Pd 介导。在本期中,Johnson 和 Stradiotto (DOI: 10.1021/acs.joc.3c01584) 以及 Szostak (DOI: 10.1021/acs.joc.4c00103) 的报告代表了涉及强键激活的交叉偶联的例子。随着更多类别的亲电试剂成为交叉偶联反应的可行偶联伙伴,需要解决化学选择性和位点选择性问题 (28,45),如 So 等人的报告 (DOI: 10.1021/acs.joc.3c02345) 所示。关于单一有机金属催化步骤的基本机理研究,例如本期 Denmark 等人的一篇文章 (DOI: 10.1021/acs.joc.3c02629),继续推动交叉偶联的进步。在图 1B 中建立规范机制的同时,出现了 Heck 和 Sonogashira 交叉偶联,表明偏离标准途径也是可能的。(1,2,19,38) 这些机理变化激发了合成化学家的创造力,导致了对经典交叉偶联范式的几次修改。例如,用简单的 C-H 亲核试剂取代预官能团亲核试剂导致了有机合成领域的一场重大革命。 (22,40) Hruszkewycz、Leitch 及其同事在本期中的贡献利用了这一主题对杂环的直接烯基化 (DOI: 10.1021/acs.joc.3c02311)。与规范机制的更剧烈偏差是由于认识到可以通过单电子转移 (SET) 化学实现交叉耦合。此类过程可能代表了目前交叉耦合中最活跃的研究领域之一,并且可以通过化学、光化学或电化学方法实现。在本期特刊中,大多数手稿都属于以激进物种为特征的主题之一,包括 Bahamonde 的 Perspectives (DOI: 10.1021/acs.joc.3c02353)、Gesmundo (DOI: 10.1021/acs.joc.3c02351) 及其同事。尽管金属催化交叉偶联反应的早期研究集中在 C(sp2)-C(sp2) 键的形成上,但最近将 C(sp3) 亲核试剂和亲电试剂成功掺入交叉偶联方法中,使研究人员能够开发出一系列广泛的策略,通过 C(sp3)–C(sp2) 或 C(sp3)–C(sp3) 构建三维有机分子) 键形成过程。(17,25,36,43) 为了规避烷基转移金属化和氧化加成的固有挑战,需要开发新的方法,将烷基有效地转移到过渡金属催化剂上。由于烷基亲核试剂和烷基亲电试剂都可以通过单电子或双电子机制掺入催化循环中,因此可以使用各种过渡金属催化剂和交叉偶联介质设想各种交叉偶联方法。 利用 SET 机制的现代策略在促进新的 C(sp3)–C(sp2) 和 C(sp3)–C(sp3) 键形成反应的开发方面特别有影响力。总的来说,这些新方法在当前期刊中得到了很好的体现,其中许多交叉偶联研究的特点是反应涉及至少一个烷基伴侣。在本期特刊中,丹麦 (DOI: 10.1021/acs.joc.4c00089) 描述了一种 Pd 催化的 C(sp3)–C(sp2) 键形成 Suzuki-Miyaura 反应,该反应涉及通过双电子机制进行金属转移的伯烷基硼酯。Noël 和 Watson 分别报告了涉及使用 Ni 催化烷基脱氨以获得烷基自由基以用于交叉偶联反应的研究。Noël (DOI: 10.1021/acs.joc.3c00859) 通过电催化实现的 SET 脱氨途径将烷基掺入 Ni 催化的交叉亲电反应中。相比之下,Watson (DOI: 10.1021/acs.joc.3c00665) 采用镍催化方法,选择性地将烷基锌亲核试剂与脱氨产生的烷基配偶体偶联。Waldvogel 和 Rousseaux 都描述了新的镍催化交叉偶联反应,其中通过使用氧化还原活性酯实现烷基自由基的产生。Waldvogel (DOI: 10.1021/acs.joc.4c00428) 结合镍催化和电催化,从氧化还原活性酯生成伯烷基自由基,用于合成西格列汀。Rousseaux (DOI: 10.1021/acs.joc.3c02354) 采用氧化还原镍催化的氧化还原活性酯和芳基碘化物交叉偶联来制备α-芳基腈。 Xiao、Shen 和 Roberts 的工作都通过过渡金属催化和可见光催化的合并,使烷基自由基参与交叉偶联反应。在 Xiao (DOI: 10.1021/acs.joc.3c02348) 的工作中,烷基锗亲核试剂通过光催化产生叔烷基自由基,并掺入随后的镍催化酰化反应中。Shen (DOI: 10.1021/acs.joc.3c02293) 将镍催化和光催化融合在立体收敛还原偶联中,能够形成高度对映体富集的苯甲醇。Roberts (DOI: 10.1021/acs.joc.3c00872) 以一种概念新颖的烷基自由基形成方法,以光催化的 Cu 介导的交叉偶联方法合并烷基脱羧和烷基脱磺酰化,以实现苄基三氟甲基化。最后,Giri (DOI: 10.1021/acs.joc.3c02548) 描述了苯乙烯二苄基化过程,该过程通过铁促进苄基自由基的形成进行。SET 通路也可用于不涉及烷基偶联伴侣的下一代交叉偶联。例如,在本期中,Li、Sun 和 Ding (DOI: 10.1021/acs.joc.4c00021) 采用电化学交叉偶联来获取联芳基产物。Kakiuchi (DOI: 10.1021/acs.joc.3c02601) 描述了电化学与钯催化相结合以实现 C-H 碘化。随着不同范式的演变,高通量实验 (HTE) 等新技术的出现,以最好地利用交叉偶联的潜力。(29,49) HTE 已成为过程和药物化学活动的中流砥柱,通过平行库合成实现快速反应优化和化学空间探索。 HTE 可用于偶联伴侣组合的反应条件的多参数优化,如 Bock 和 Denmark 在本期的报告中所示 (DOI: 10.1021/acs.joc.4c00089)。或者,HTE 可用于同时筛选各种偶联配偶体的反应条件,正如 Gesmundo 及其同事在 AbbVie (DOI: 10.1021/acs.joc.3c02351) 和 Leitch (DOI: 10.1021/acs.joc.3c02311) 与 GSK 的研究人员合作所讨论的那样。后一种筛选通常揭示一组反应条件,这些条件共同使人们能够获得比任何单一方法更广泛的化学空间。虽然 HTE 被用于探索热反应已有十多年,但其用于光化学和电化学转化的采用直到最近几年才浮出水面。正如 Gesmundo 等人 (DOI: 10.1021/acs.joc.3c02351) 的 Perspective 中所示,HTE 在光化学转化中的广泛采用需要对设备和反应条件进行系统探索,以确定用户友好、稳健的工艺,并通过广泛的偶联伙伴实现高成功率。随着电化学 HTE 设备的出现,可能需要类似的系统方法才能在工业界和学术界无缝采用高通量电化学转化。虽然 HTE 使化学空间的快速探索成为可能,但筛选仍然需要大量时间和资源。因此,前瞻性方法需要使用 HTE 数据来构建和应用用于合成转换的预测 ML 模型。(32) 交叉耦合变换的最早范式激发了一个多世纪以来跨越不同方向的创新。 即使现代化学家继续推进更成熟的交叉偶联研究主题,这些主题也逐渐分支到新的方向。与此同时,过去几十年见证了 HTE 等技术的创新,以加速学术界和工业界对下一代交叉耦合的优化和发现,通常是通过合作。我们预计未来的创新,包括数据科学和新机理流形的发现,将继续扩大交叉偶联化学的多样性。本文引用了其他 49 种出版物。有关最近的相关评论,请参阅:本文尚未被其他出版物引用。
更新日期:2024-11-15
中文翻译:
从成熟到新兴:交叉偶联反应的演变
本文是 Next-Generation Cross-Coupling Chemistry 特刊的一部分。在 C 原子和生物相关元素(如 C、O、N、S 和 P(等)之间构建键是合成有机化学家的基本目标。尽管 C-C 键形成反应(例如,羟醛化学、格氏试剂的反应)取得了早期进展,但在 1960 年代,一个关键挑战仍然存在:在两个 C(sp2) 原子之间有效构建 C-C 键(图 1A)。到 1970 年代后期,多位化学家的工作集中在一种解决方案上,该解决方案涉及来自主要族有机金属物种 [C(sp2)–M;M = B、Sn、Zn 或 Mg] 与芳基亲电试剂 [C(sp2)–X;X = I、Br、Cl、OTf] 在 Ni 或 Pd 催化剂存在下。(1−8) 随着这些发现,出现了形成化学键的新范式。C-C 交叉偶联反应的重要性和影响使其与酰胺键形成和亲核取代一起在合成领域一跃成为受人尊敬的地位。(9) 2010 年,诺贝尔化学奖颁发给了引领这场革命的贡献者,即铃木明、根岸荣一和理查德 F. 赫克。(10) 图 1.(A) Pd 催化的交叉偶联反应。(B) 涉及有机金属亲核试剂的交叉偶联的经典机制。尽管术语“交叉偶联”的起源通常与 Suzuki-Miyaura、Stille、Kumada 和 Negishi 反应(以其发明者命名)的发展有关,但早期的例子存在于 Glaser、(11)、Ullman、(12) 或 Kharasch (13) 涉及有机金属试剂和芳基卤化物之间的反应由过渡金属盐催化。 然而,低选择性和产量以及缺乏对机械的理解限制了此类策略的广泛采用。另一方面,Pd 催化的交叉偶联反应提供了高产率、拓宽了底物范围以及分离和研究稳定有机金属中间体的可能性。多年来,文献中出现了许多过渡金属催化交叉偶联反应的机理研究。基于这些研究,社区达成了一项协议,即涉及有机金属亲核试剂的交叉偶联的催化歧管包括三个主要的有机金属步骤:氧化加成、金属转移和还原消除(图 1B)。尽管每个机理步骤都有其自身的复杂性和复杂性,但这一总体情况为有机化学家提供了理解和预测反应性、选择性和其他特征的指导原则,以推进交叉偶联领域。从交叉偶联策略的不断发展来看,令人眼花缭乱的机制、试剂类别和产品结构的多样性是现代交叉偶联的特点。由于这些反应偏离了规范交叉偶联的传统特征,因此术语“交叉偶联”已经演变为代表一种合成哲学,而不是机理或反应物结合的定义。(14) 从一般意义上讲,交叉偶联反应可以简单地描述为将两个(或多个)构建单元合并在一起以构建新的键。这些反应由第三方介导,第三方是传统上是过渡金属催化剂的实体,但偶尔会非常不同(例如,光子,(15) 一个主要的族元素 (16))。 不能轻易归入定义明确的不同类别的反应通常可以很好地纳入更广泛的“交叉偶联”框架。受本期特刊中创意报告的启发,我们在这里尝试确定交叉耦合领域的一些研究主题。在图 2 中,沿着时间线的左侧,标记了一些点以指示每个主题的开创性出版物。(1,3,10−12,17−33)在更模糊的情况下,根据产物与规范交叉偶联的产物之间的相似性来选择具有里程碑意义的出版物。例如,Périchon 在 1993 年合成的联芳基被选为代表电化学交叉偶联时代的开始。(24) 对于许多主题来说,多年甚至几十年后,关于该主题的相对一连串活动才开始出现在文献中。时间轴右侧的点表示一些出版物,这些出版物导致了主题流行度的拐点期。(2,4−8,13,34−48)为简洁起见,我们选择了最少数量的例子,但这份关键贡献清单肯定是不完整的,许多其他论文代表了研究领域内的重要进步。本期特刊中的许多报告都属于时间轴中说明的一个或多个研究主题。其他文章,如 Li 和 Sun (DOI: 10.1021/acs.joc.3c02417)、Lv 和 Li (DOI: 10.1021/acs.joc.3c00744)、Shenvi (DOI: 10.1021/acs.joc.4c00260) 及其同事的文章,进一步扩展并强调了交叉耦合作为一种综合哲学的想法。图 2.交叉偶联 (XC) 领域选定研究主题的时间表:开创性工作(左侧)和近似拐点(右侧)。 虽然交叉偶联反应最初主要以芳基碘化物和溴化物的使用为主,但希望扩大范围以包括更具挑战性的亲电试剂(如 Ar-Cl)和酚类衍生物(如 Ar-OT 和 Ar-OM),这激发了配体设计和 Ni 催化交叉偶联的创新。自从 Wenkert 的原始报告证明了 Ni 催化的 Kumada 与 Ar-OMe 和 Ar-OCOR 的交叉偶联 (20) 以来,已经报道了多种使用具有挑战性的亲电试剂的方法,通常由高亲核性 Ni 催化剂 (44) 或由大体积配体支持的 Pd 介导。在本期中,Johnson 和 Stradiotto (DOI: 10.1021/acs.joc.3c01584) 以及 Szostak (DOI: 10.1021/acs.joc.4c00103) 的报告代表了涉及强键激活的交叉偶联的例子。随着更多类别的亲电试剂成为交叉偶联反应的可行偶联伙伴,需要解决化学选择性和位点选择性问题 (28,45),如 So 等人的报告 (DOI: 10.1021/acs.joc.3c02345) 所示。关于单一有机金属催化步骤的基本机理研究,例如本期 Denmark 等人的一篇文章 (DOI: 10.1021/acs.joc.3c02629),继续推动交叉偶联的进步。在图 1B 中建立规范机制的同时,出现了 Heck 和 Sonogashira 交叉偶联,表明偏离标准途径也是可能的。(1,2,19,38) 这些机理变化激发了合成化学家的创造力,导致了对经典交叉偶联范式的几次修改。例如,用简单的 C-H 亲核试剂取代预官能团亲核试剂导致了有机合成领域的一场重大革命。 (22,40) Hruszkewycz、Leitch 及其同事在本期中的贡献利用了这一主题对杂环的直接烯基化 (DOI: 10.1021/acs.joc.3c02311)。与规范机制的更剧烈偏差是由于认识到可以通过单电子转移 (SET) 化学实现交叉耦合。此类过程可能代表了目前交叉耦合中最活跃的研究领域之一,并且可以通过化学、光化学或电化学方法实现。在本期特刊中,大多数手稿都属于以激进物种为特征的主题之一,包括 Bahamonde 的 Perspectives (DOI: 10.1021/acs.joc.3c02353)、Gesmundo (DOI: 10.1021/acs.joc.3c02351) 及其同事。尽管金属催化交叉偶联反应的早期研究集中在 C(sp2)-C(sp2) 键的形成上,但最近将 C(sp3) 亲核试剂和亲电试剂成功掺入交叉偶联方法中,使研究人员能够开发出一系列广泛的策略,通过 C(sp3)–C(sp2) 或 C(sp3)–C(sp3) 构建三维有机分子) 键形成过程。(17,25,36,43) 为了规避烷基转移金属化和氧化加成的固有挑战,需要开发新的方法,将烷基有效地转移到过渡金属催化剂上。由于烷基亲核试剂和烷基亲电试剂都可以通过单电子或双电子机制掺入催化循环中,因此可以使用各种过渡金属催化剂和交叉偶联介质设想各种交叉偶联方法。 利用 SET 机制的现代策略在促进新的 C(sp3)–C(sp2) 和 C(sp3)–C(sp3) 键形成反应的开发方面特别有影响力。总的来说,这些新方法在当前期刊中得到了很好的体现,其中许多交叉偶联研究的特点是反应涉及至少一个烷基伴侣。在本期特刊中,丹麦 (DOI: 10.1021/acs.joc.4c00089) 描述了一种 Pd 催化的 C(sp3)–C(sp2) 键形成 Suzuki-Miyaura 反应,该反应涉及通过双电子机制进行金属转移的伯烷基硼酯。Noël 和 Watson 分别报告了涉及使用 Ni 催化烷基脱氨以获得烷基自由基以用于交叉偶联反应的研究。Noël (DOI: 10.1021/acs.joc.3c00859) 通过电催化实现的 SET 脱氨途径将烷基掺入 Ni 催化的交叉亲电反应中。相比之下,Watson (DOI: 10.1021/acs.joc.3c00665) 采用镍催化方法,选择性地将烷基锌亲核试剂与脱氨产生的烷基配偶体偶联。Waldvogel 和 Rousseaux 都描述了新的镍催化交叉偶联反应,其中通过使用氧化还原活性酯实现烷基自由基的产生。Waldvogel (DOI: 10.1021/acs.joc.4c00428) 结合镍催化和电催化,从氧化还原活性酯生成伯烷基自由基,用于合成西格列汀。Rousseaux (DOI: 10.1021/acs.joc.3c02354) 采用氧化还原镍催化的氧化还原活性酯和芳基碘化物交叉偶联来制备α-芳基腈。 Xiao、Shen 和 Roberts 的工作都通过过渡金属催化和可见光催化的合并,使烷基自由基参与交叉偶联反应。在 Xiao (DOI: 10.1021/acs.joc.3c02348) 的工作中,烷基锗亲核试剂通过光催化产生叔烷基自由基,并掺入随后的镍催化酰化反应中。Shen (DOI: 10.1021/acs.joc.3c02293) 将镍催化和光催化融合在立体收敛还原偶联中,能够形成高度对映体富集的苯甲醇。Roberts (DOI: 10.1021/acs.joc.3c00872) 以一种概念新颖的烷基自由基形成方法,以光催化的 Cu 介导的交叉偶联方法合并烷基脱羧和烷基脱磺酰化,以实现苄基三氟甲基化。最后,Giri (DOI: 10.1021/acs.joc.3c02548) 描述了苯乙烯二苄基化过程,该过程通过铁促进苄基自由基的形成进行。SET 通路也可用于不涉及烷基偶联伴侣的下一代交叉偶联。例如,在本期中,Li、Sun 和 Ding (DOI: 10.1021/acs.joc.4c00021) 采用电化学交叉偶联来获取联芳基产物。Kakiuchi (DOI: 10.1021/acs.joc.3c02601) 描述了电化学与钯催化相结合以实现 C-H 碘化。随着不同范式的演变,高通量实验 (HTE) 等新技术的出现,以最好地利用交叉偶联的潜力。(29,49) HTE 已成为过程和药物化学活动的中流砥柱,通过平行库合成实现快速反应优化和化学空间探索。 HTE 可用于偶联伴侣组合的反应条件的多参数优化,如 Bock 和 Denmark 在本期的报告中所示 (DOI: 10.1021/acs.joc.4c00089)。或者,HTE 可用于同时筛选各种偶联配偶体的反应条件,正如 Gesmundo 及其同事在 AbbVie (DOI: 10.1021/acs.joc.3c02351) 和 Leitch (DOI: 10.1021/acs.joc.3c02311) 与 GSK 的研究人员合作所讨论的那样。后一种筛选通常揭示一组反应条件,这些条件共同使人们能够获得比任何单一方法更广泛的化学空间。虽然 HTE 被用于探索热反应已有十多年,但其用于光化学和电化学转化的采用直到最近几年才浮出水面。正如 Gesmundo 等人 (DOI: 10.1021/acs.joc.3c02351) 的 Perspective 中所示,HTE 在光化学转化中的广泛采用需要对设备和反应条件进行系统探索,以确定用户友好、稳健的工艺,并通过广泛的偶联伙伴实现高成功率。随着电化学 HTE 设备的出现,可能需要类似的系统方法才能在工业界和学术界无缝采用高通量电化学转化。虽然 HTE 使化学空间的快速探索成为可能,但筛选仍然需要大量时间和资源。因此,前瞻性方法需要使用 HTE 数据来构建和应用用于合成转换的预测 ML 模型。(32) 交叉耦合变换的最早范式激发了一个多世纪以来跨越不同方向的创新。 即使现代化学家继续推进更成熟的交叉偶联研究主题,这些主题也逐渐分支到新的方向。与此同时,过去几十年见证了 HTE 等技术的创新,以加速学术界和工业界对下一代交叉耦合的优化和发现,通常是通过合作。我们预计未来的创新,包括数据科学和新机理流形的发现,将继续扩大交叉偶联化学的多样性。本文引用了其他 49 种出版物。有关最近的相关评论,请参阅:本文尚未被其他出版物引用。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号