Advanced Synthesis & Catalysis ( IF 4.4 ) Pub Date : 2024-11-13 , DOI: 10.1002/adsc.202401189 Daiki Oka, Tomomi Ohtsuka, Ken-ichi Takao, Akihiro Ogura
|
Tetrahydro-β-carbolines (THCs) can be found in many biologically valuable natural compounds and medicines (Figure 1). Typical examples are the neurogenesis stimulator, pinoline;1 the antihypertensive, ajmalicine;2 and the antihypertensive- and depression-model preparation reagent, reserpine.3 Thus, efficient methods for preparing a wide variety of THCs are required to advance exploratory research for lead compounds.

Biologically active compounds with a THC moiety.
Among the various methods for synthesizing THCs,4-7 the most straightforward approach is using the corresponding tryptamine, usually via a Pictet-Spengler reaction (Scheme 1a).8-11 The appropriate aldehyde and the tryptamine are condensed to afford the intermediate imine, which is treated with an acid catalyst to induce carbon-carbon bond formation, providing THCs. Equally valuable is the Bischler-Napieralski approach,12, 13 in which the tryptamine is converted to an amide via condensation, and dehydroxylative activation of the amide moiety followed by hydrogenation affords the desired THC. However, from a synthetic perspective, the palette of tryptamines is not large enough. For example, among readily available 20 substitution patterns on an indole moiety, only the parent tryptamine and 5-methoxy derivative are available at a cost below 100 USD/g (as of September 2024).

Synthesis of THCs.
Another well-established procedure is the Fischer indole synthesis (Scheme 1b).14-16 An aryl hydrazine and α-aminoketone are condensed to give the hydrazone, which is isomerized by acid treatment to the corresponding enamine. The key carbon-carbon bond is formed by spontaneous [3,3]-sigmatropic rearrangement of the enamine. The major drawback is the involvement of an unstable α-aminoketone;17 therefore, many examples of this reaction use an α-oxolactam instead of an α-aminoketone.
To build THCs from a simpler unit, simultaneous construction of the pyrrole ring and piperidine ring is attractive (Scheme 1c). Bondzic and Eilbracht conducted sequential hydroformylation of dihydropyrrole, Fischer-type sigmatropic rearrangement, and bond migration to afford THCs.18 Fujii and Ohno synthesized an aryl propargyl amine intermediate by a copper-mediated multicomponent reaction and performed an elegant one-pot cyclization to yield THCs.19 Similar aryl propargyl amines were also used by other groups for THC synthesis using iodine, gold, or palladium.20-22
We recently reported an environmentally benign synthesis of 3-methylindoles by sequential sigmatropic rearrangements (Scheme 1d),23 in which hydrogen peroxide-mediated oxidation of an allyl aniline was followed by spontaneous [2,3]-Meisenheimer rearrangement24 to afford N-allyloxy anilines. Subsequent olefin isomerization gave N-vinyloxy aniline, which underwent aza-oxa-Cope rearrangement and aromatization with water expulsion to give indoles. The whole sequence requires no neutralization because the waste products generated are only water and trichloroacetamide.
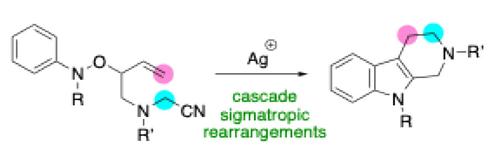
This allyloxyaniline isomerization was developed further as a strategy for heterocycle construction, and we conceived a synthesis of THCs via cascade [3,3]-sigmatropic rearrangements.25-40 Herein, we combined an aza-Cope rearrangement and aza-oxa-Cope rearrangement to construct the THC skeleton in good yield under mild reaction conditions (Scheme 1e).
We expected that the Overman iminium cation41 (Scheme 1e, bottom left) would undergo an aza-Cope rearrangement to afford N-vinyloxyaniline (bottom center), and then an aza-oxa Cope rearrangement23, 42-55 would occur to form the second carbon-carbon bond. Aromatization accompanied with water expulsion would form the indole ring, and finally Pictet-Spengler cyclization would close the last ring to afford a THC.
Our substrate preparation is shown in Scheme 2. Aniline 1 a was alkylated with 2 a to give 3 a. N-Oxidation was performed with m-CPBA, and [2,3]-sigmatropic rearrangement followed immediately to afford the corresponding allyloxyaniline. Electron-withdrawing groups on aniline nitrogen atom (e. g., Piv) did not afford N-oxidation or sigmatropic rearrangement product, due to reduced electron density on nitrogen atom. The Boc group was removed with a combination of activator TMSOTf and trap reagent anisole.56 Using a strong Brønsted acid (e. g., trifluoroacetic acid) caused decomposition. The synthesis with various substituents proceeded uneventfully under identical conditions to afford corresponding 4 in good yield (see SI).

Synthesis of reaction substrate 5 a.
We initially expected that condensation of secondary amine 4 with an appropriate aldehyde under acidic conditions would form an iminium cation directly in situ, which would trigger the aza-Cope/aza-oxa-Cope rearrangement pathway. However, the reaction resulted in decomposition, seemingly due to N−O bond scission, and no aza-Cope reaction products were detected.
Then, we decided to use a cyanomethyl group as a precursor to the iminium cation.57 Thus, 4 a was converted to cyanomethylamine 5 a, which was treated with a silver source, expecting that the high affinity of the silver cation would facilitate the elimination of the cyano group to generate the iminium species. The original conditions reported by Overman, of silver nitrate in ethanol (Table 1, entry 1),57 afforded desired THC 6, although in trace amounts due to decomposition during the reaction. When excess silver nitrate to ensure reaction completion was heated in chloroform, the yield increased dramatically to 46% of 6 a (entry 2). Then, we screened silver sources (entries 3–6). Although silver acetate resulted in no reaction (entry 3), other silver salts with a stable counter anion led to total consumption of 5 a, and silver trifluoroacetate performed best (entry 4), giving a yield as high as 89%. Among the solvents tested, chloroform remained the best (entries 7–9, more trials in SI). Finally, the equivalents of the reagent were decreased. The reaction reached completion with 1.25 equiv. of silver trifluoroacetate to afford 6 a in comparable yield at a concentration of 0.2 M (entry 10). However, the addition of molecular sieves to absorb water generated during the reaction did not improve the yield (entry 11). Attempts to render the silver reagent catalytic, i. e., by using TMSOTf as a cyano group scavenger and phenanthroline as a ligand to increase silver ion solubility, have resulted in partial success so far (entry 12). To show practicality of the reaction, we repeated the experiment with the optimized conditions in gram scale, and obtained as high as 87% yield (entry 13). Notably, the reaction involved seven bond cleavages and four new bond formations,58 and yield per bond connection or disconnection was as high as 99%.

- [a] conducted with 5 s (see SI for detailed structure), 20 h. [b] 0.2 M. [c] MS 4 A was added. [d] 1.25 eq. TMSOTf was added, 4 h. [e] 1 g scale.
Having established the optimum conditions, we investigated the substrate scope (Table 2). The reaction proceeded in good to medium yield when the aryl unit was unsubstituted or was substituted with an electron-donating alkyl group or halogen atom at the para position (6 b to 6 f). Introducing a more electron-donating group (e. g., a methoxy group) to the substrate 6 failed because the parent amine 4 was too unstable. When the para substituent was electron-withdrawing, the reaction required 2.5 equiv. silver salt, and the yield was lower (48% for 6 g and 39% for 6 h). In particular, cyano substitution led to a complete loss of yield even with a larger amount of silver trifluoroacetate (6 i) because of competitive complexation of the silver cation at the cyano substituent and possible nucleophilic attack on this position. Some of these results were improved if 2,6-lutidine was added as a base (57% for 6 h and 17% for 6 i).

- [a] 2.5 eq. AgOTFA. [b] 1.25 eq. 2,6-lutidine. [c] 2.5 h.
Other substitution patterns were examined. meta Substitution led to a mixture of isomers, 6 j and 6 j′, owing to an uncontrolled aza-oxa-Cope reaction. In contrast, ortho substitution gave single product 6 k in good yield. The naphthylamine-derived substrate gave 6 l in modest yield, which might be attributed to complexation of silver salt to extended aromatic system.59
Substitution on the nitrogen atoms was investigated. Various alkyl groups on aniline nitrogen, such as methyl, benzyl, ethoxycarbonylmethyl, and bulky isopropyl groups, were tolerated (6 m–6 p). The benzyl group of 6 n could be removed under Birch reduction conditions (see SI). Structurally interesting tetracycle 6 q was obtained when tetrahydroquinoline was used as the parent aniline. Phenyl substitution was also tolerated to provide 6 r. The other nitrogen atom could bear an ethyl group or benzyl group (6 s and 6 t), although 6 t had a slightly lower yield.
To clarify the mechanism, we performed crossover reactions to confirm whether the aza-oxa-Cope rearrangement was intramolecular or intermolecular, and whether the final Pictet–Spengler reaction involved an aldehyde exchange. First, the reaction was performed in the presence of excess external aldehyde to confirm the Pictet-Spengler step (Figure 2a). The reaction gave only intramolecular product 6 a, and no trace of crossover product 7 or 8, which would be formed by iminium exchange before or after the aza-Cope rearrangement, respectively, was observed by NMR or mass spectrometry. When n-butanal was used, the yield decreased, which was attributed to competing hydrolysis due to water contamination. Thus, no hydrolysis of the iminium intermediate occurred before the Pictet-Spengler reaction in the mechanism. Next, we conducted a crossover reaction using equimolar amounts of 5 c and 5 t to figure out the aza-oxa-Cope rearrangement mechanism (Figure 2b). The reaction afforded only 6 c and 6 t, and no trace of crossover product 6 u or 6 v was detected. These experiments confirmed that the reaction proceeded entirely via the intramolecular pathway after cyano group extrusion.

Mechanistic analysis. (a) Crossover experiment with external aldehyde. AgOTFA (1.25 equiv), aldehyde (4.0 equiv), CHCl3, 60 °C. (b) Crossover experiment with two substrates. 5 c and 5 t (1.0 equiv), AgOTFA (2.5 equiv), CHCl3, 60 °C. (c) NMR time-course analysis. The reaction was performed inside an NMR sample tube with CDCl3 as the solvent and 1,3,5-trimethoxy benzene as the internal standard. (d) Yield versus Hammett constant. Yield without 2,6-lutidine was used for p-CF3. (e) DFT calculation.
We performed NMR time course analysis, expecting to observe any intermediary compound (Figure 2c). The reaction proceeded slower than in the flask due to mixing inefficiency. However, we could only see peaks for starting molecule 5 a and product 6 a, and no other signals from intermediary compounds were detected. Thus, as we saw during previous metal-catalyzed indole synthesis,23 not only aza-Cope rearrangement57 but also aza-oxa-Cope rearrangement was fast42-49 compared with the preceding elimination of the cyano group.
The reaction yield was generally low when an electron-withdrawing substituent was introduced on the aromatic ring or when the aliphatic nitrogen carried a bulky group. This was attributed to the final Pictet-Spengler step, because higher electron density on the aromatic ring or a less-bulky iminium cation are favourable for this reaction. A plot of the yield versus Hammett's substituent constant showed good correspondence (Figure 2d). A slow Pictet–Spengler reaction led to the decomposition of the intermediate iminium cation, which lowered the yield.
To support the proposed reaction mechanism, a density functional theory (DFT) calculation of the key aza-Cope/aza-oxa-Cope rearrangements was performed (Figure 2e).60 The calculation started from initial iminium cation SM. The first aza-Cope reaction was almost barrierless, and led to enol intermediate IM1-cis via TS1-cis, or IM1-trans via TS1-trans. Interestingly, IM1-cis was more stable than IM1-trans by 2.6 kcal/mol. The following aza-oxa-Cope rearrangement could proceed via the chair form or boat form. Among the possible transition states, TS2a-trans and TS2b-cis were almost equally stable, due to the reduced steric repulsion between the aromatic ring and the side chain. The energy barrier was low enough for the reaction to reach completion smoothly under the reaction conditions (ca. 60 °C),61 which was observed in the NMR time course analysis (Figure 2c). Although the reaction appeared to occur via the TS1-cis/IM1-cis/TS2b-cis pathway, the route via TS2a-trans could also be a competing pathway via equilibration under mild heating.
In conclusion, we developed a method for constructing THCs from readily available anilines. The reaction occurred via a cascade of aza-Cope/aza-oxa-Cope/Pictet-Spengler reactions, and involved seven bond disconnections and four bond formations with just an equimolar amount of reagent. The reaction mechanism was supported by a mechanistic analysis, including crossover experiments and a DFT calculation. We believe this method offers an attractive alternative to tryptamine- or arylhydrazine-based THC synthesis. We are currently investigating further applications of this strategy for heterocycle synthesis.
Experimental Section
General Procedure for Conversion of 5–6
To a stirred solution of 5 (1 eq.) in CHCl3 (0.2 M) was added silver trifluoroacetate (1.25 eq.) under Ar atmosphere. The mixture was stirred at 60 °C for 30 min. The precipitated solids were removed by filtration through a pad of Celite and washed well with CH2Cl2. The combined filtrate and washings were quenched with 1 M aqueous NaOH, diluted with H2O and extracted with CH2Cl2 (3 times). The combined extracts were dried and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel.
Detailed procedures and analytical data for all new compounds are given in the Supporting Information.
Acknowledgments
This research was supported by Fujimori Science and Technology Foundation.
中文翻译:
通过级联西格马形重排合成四氢-β-咔啉
四氢β-咔啉 (THC) 存在于许多具有生物价值的天然化合物和药物中(图 1)。典型的例子是神经发生刺激剂 pinoline;1 抗高血压药 ajmalicine;2 和抗高血压和抑郁症模型制备试剂利血平。3 因此,需要有效的方法来制备各种 THC,以推进铅化合物的探索性研究。
在图窗查看器PowerPoint 中打开
具有 THC 部分的生物活性化合物。
在合成 THC 的各种方法中,4-7 最直接的方法是使用相应的色胺,通常通过 Pictet-Spengler 反应(方案 1a)。8-11 适当的醛和色胺被缩合得到中间体亚胺,该亚胺用酸催化剂处理以诱导碳-碳键形成,从而提供 THC。同样有价值的是 Bischler-Napieralski 方法,12, 13 其中色胺通过缩合转化为酰胺,酰胺部分的脱羟基活化,然后氢化提供所需的 THC。然而,从合成的角度来看,色胺的调色板还不够大。例如,在一个吲哚部分上现成的 20 种取代模式中,只有母体色胺和 5-甲氧基衍生物的成本低于 100 美元/克(截至 2024 年 9 月)。
在图窗查看器PowerPoint 中打开
四氢大麻酚的合成。
另一个成熟的程序是 Fischer 吲哚合成(方案 1b)。14-16 芳基肼和 α-氨基酮缩合得到腙,腙通过酸处理异构化成相应的烯胺。关键的碳-碳键是由烯胺的自发 [3,3]-σ 重排形成的。主要缺点是不稳定的 α-氨基酮的参与;17 因此,该反应的许多例子使用 α-氧内酰胺而不是 α-氨基酮。
为了从更简单的单元构建 THC,同时构建吡咯环和哌啶环很有吸引力(方案 1c)。Bondzic 和 Eilbracht 进行了二氢吡咯的连续加氢甲酰化、Fischer 型 σ 重排和键迁移,以获得 THC.18 Fujii 和 Ohno 通过铜介导的多组分反应合成了芳基炔丙基胺中间体,并进行了优雅的一锅法环化以产生 THC.19 类似的芳基丙炔胺也被其他小组用于使用碘合成 THC, 金或钯。20-22 元
我们最近报道了一种通过连续 σ 重排(方案 1d)合成 3-甲基吲哚的环境良性合成(方案 1d)23,其中过氧化氢介导的烯丙基苯胺氧化后自发 [2,3]-Meisenheimer 重排24 得到 N-烯丙氧基苯胺。随后的烯烃异构化得到 N-乙烯氧基苯胺,它经历氮杂-氧杂-Cope 重排和芳构化,用水排出得到吲哚。整个序列不需要中和,因为产生的废物只有水和三氯乙酰胺。
这种烯丙氧基苯胺异构化作为杂环构建的策略得到进一步发展,我们构想了通过级联 [3,3] -σ 促性重排合成 THC。25-40 在此,我们结合了 aza-Cope 重排和 aza-oxa-Cope 重排,以在温和的反应条件下以良好的产率构建 THC 骨架(方案 1e)。
我们预计 Overman 亚胺阳离子41(方案 1e,左下)将经历 aza-Cope 重排以提供 N-乙烯氧基苯胺(中下),然后发生 aza-oxa Cope 重排23、42-55 以形成第二个碳-碳键。芳构化伴随着水排出会形成吲哚环,最后 Pictet-Spengler 环化将关闭最后一个环以获得 THC。
我们的底物制备如方案 2 所示。苯胺 1 a 与 2 a 烷基化,得到 3 a。用 m-CPBA 进行 N-氧化,然后立即进行 [2,3]-σ-σ-偏斜重排,得到相应的烯丙氧基苯胺。由于氮原子上的电子密度降低,苯胺氮原子(例如 Piv)上的吸电子基团不产生 N 氧化或 σ 重排产物。用活化剂 TMSOTf 和捕集试剂苯甲醚的组合去除 Boc 组。56 使用强 Brønsted 酸(例如三氟乙酸)会导致分解。在相同的条件下,与各种取代基的合成顺利进行,以获得相应的 4 的良好产率(参见 SI)。
在图窗查看器PowerPoint 中打开
反应底物的合成 5 a.
我们最初预计仲胺 4 在酸性条件下与适当的醛缩合会直接在原位形成亚胺阳离子,这将触发 aza-Cope/aza-oxa-Cope 重排途径。然而,该反应导致分解,似乎是由于 N-O 键断裂,并且没有检测到 aza-Cope 反应产物。
然后,我们决定使用氰甲基作为亚胺阳离子的前驱体。57 因此,4 a 转化为氰甲胺 5 a,用银源处理,预期银阳离子的高亲和力将促进消除氰基以生成亚胺物质。Overman 报告的乙醇中硝酸银的原始条件(表 1,条目 1),57 提供了所需的 THC 6,尽管由于反应过程中的分解而微量。当过量的硝酸银在氯仿中加热以确保反应完成时,产率急剧提高到 6 A 的 46%(条目 2)。然后,我们筛选了银源(条目 3-6)。虽然乙酸银没有反应(条目 3),但其他具有稳定反阴离子的银盐导致总消耗量为 5 a,而三氟乙酸银表现最好(条目 4),产率高达 89%。在测试的溶剂中,氯仿仍然是最好的(条目 7-9,SI 中的更多试验)。最后,试剂的当量减少。用 1.25 当量的三氟乙酸银完成反应,在 0.2 M 的浓度下得到 6 a 的相当产量(条目 10)。然而,添加分子筛以吸收反应过程中产生的水并没有提高产量(条目 11)。迄今为止,尝试使银试剂具有催化作用,即使用 TMSOTf 作为氰基清除剂,使用菲咯啉作为配体来提高银离子溶解度,已部分成功(条目 12)。 为了显示反应的实用性,我们在克级的最佳条件下重复实验,并获得了高达 87% 的产率(条目 13)。值得注意的是,该反应涉及 7 个键裂解和 4 个新键形成,58 每个键连接或断开的产量高达 99%。
[a] 以 5 秒进行(详细结构见 SI),20 小时。[b] 0.2 M. [c] 添加了 MS 4 A。[d] 1.25 等式 加入 TMSOTf,4 小时。[e] 1 g 刻度。
确定了最佳条件后,我们研究了衬底范围(表 2)。当芳基单元未被取代或在对位(6 b 至 6 f)被供电子烷基或卤素原子取代时,反应以良好至中等产率进行。将更多的电子供体基团(例如甲氧基)引入底物 6 失败,因为母胺 4 太不稳定。当对位取代基为吸电子时,反应需要 2.5 当量银盐,并且产率较低(6 g 为 48%,6 h 为 39%)。特别是,氰基取代导致即使使用大量三氟乙酸银 (6 i) 也会导致产率完全损失,因为银阳离子在氰基取代基处的竞争性络合和对该位置的可能亲核攻击。如果添加 2,6-lutidine 作为碱,其中一些结果会得到改善(6 h 为 57%,6 i 为 17%)。
表 2. THC 合成的底物范围。
[a] 2.5 方程AgOTFA.[b] 1.25 方程2,6-二明二胺。[c] 2.5 小时
检查了其他替代模式。元取代导致异构体 6 j 和 6 j′ 的混合物,由于不受控制的 aza-oxa-Cope 反应。相比之下,邻位取代以良好的产率获得 6 k 的单个产物。萘胺衍生的底物以适度的产量产生 6 l,这可能归因于银盐与扩展芳香族系统的络合。59 元
研究了氮原子上的取代。苯胺氮上的各种烷基,如甲基、苄基、乙氧基羰基甲基和庞大的异丙基,是耐受的 (6 m–6 p)。在 Birch 还原条件下可以去除 6 n 的苄基(参见 SI)。当使用四氢喹啉作为母体苯胺时,获得了结构上有趣的四环 6 q。苯基取代也被耐受以提供 6 r。另一个氮原子可以承载乙基或苄基(6 s 和 6 t),尽管 6 t 的产率略低。
为了阐明机制,我们进行了交叉反应以确认 aza-oxa-Cope 重排是分子内还是分子间,以及最终的 Pictet-Spengler 反应是否涉及醛交换。首先,在过量的外醛存在下进行反应,以确认 Pictet-Spengler 步骤(图 2a)。反应仅得到分子内产物 6 a,没有通过 NMR 或质谱法分别观察到在 aza-Cope 重排之前或之后通过亚胺交换形成的痕量交叉产物 7 或 8。当使用正丁醛时,产量下降,这归因于水污染导致的竞争性水解。因此,在机理中的 Pictet-Spengler 反应之前没有发生亚胺中间体的水解。接下来,我们使用 5 c 和 5 t 的等摩尔量进行交叉反应,以找出 aza-oxa-Cope 重排机制(图 2b)。反应仅提供 6 c 和 6 t,未检测到痕量交叉产物 6 u 或 6 v。这些实验证实,反应完全通过氰基挤出后的分子内途径进行。
在图窗查看器PowerPoint 中打开
机理分析。(a) 与外醛的交叉实验。AgOTFA(1.25 当量)、醛(4.0 当量)、CHCl 3、60 °C。(b) 两种底物的交叉实验。5 c 和 5 t(1.0 当量),AgOTFA(2.5 当量),CHCl 3,60 °C。(c) NMR 时程分析。反应在 NMR 样品管内进行,以 CDCl3 为溶剂,以 1,3,5-三甲氧基苯为内标。(d) 产量与哈米特常数。不含 2,6-lutidine 的产量用于 p-CF 3。(e) DFT 计算。
我们进行了 NMR 时程分析,期望观察到任何中间化合物(图 2c)。由于混合效率低下,反应进行得比在培养瓶中慢。然而,我们只能看到起始分子 5 a 和产物 6 a 的峰,没有检测到来自中间化合物的其他信号。因此,正如我们在先前的金属催化吲哚合成中所看到的,23 不仅 aza-Cope 重排57 而且 aza-oxa-Cope 重排也很快42-49 与先前的氰基消除相比。
当在芳香环上引入吸电子取代基或脂肪族氮携带大体积基团时,反应产率通常较低。这归因于最后的 Pictet-Spengler 步骤,因为芳香环上较高的电子密度或体积较小的亚胺阳离子有利于该反应。产率与 Hammett 取代基常数的关系图显示出良好的对应关系(图 2d)。缓慢的 Pictet-Spengler 反应导致中间体亚胺阳离子分解,从而降低了产率。
为了支持所提出的反应机理,对关键的 aza-Cope/aza-oxa-Cope 重排进行了密度泛函理论 (DFT) 计算(图 2e)。60 计算从初始亚胺阳离子 SM 开始。第一个 aza-Cope 反应几乎是无屏障的,并通过 TS1-cis 导致烯醇中间体 IM1-cis,或通过 TS1-trans 导致 IM1-trans。有趣的是,IM1-cis 比 IM1-trans 更稳定 2.6 kcal/mol。以下 aza-oxa-Cope 重排可以通过 chair form 或 boat form 进行。在可能的过渡态中,TS2a-trans 和 TS2b-cis 几乎同样稳定,因为芳香环和侧链之间的空间排斥力降低。能垒足够低,使反应在反应条件(约 60 °C)下顺利完成,61 在 NMR 时程分析中观察到(图 2c)。尽管反应似乎是通过 TS1-cis/IM1-cis/TS2b-cis 途径发生的,但通过 TS2a-trans 的途径也可能是在温和加热下通过平衡的竞争途径。
总之,我们开发了一种用现成的苯胺构建 THC 的方法。该反应通过 aza-Cope/aza-oxa-Cope/Pictet-Spengler 反应的级联反应发生,涉及 7 个键断开和 4 个键形成,只需等摩尔量的试剂即可。反应机理得到了机理分析的支持,包括交叉实验和 DFT 计算。我们相信这种方法为基于色胺或芳肼的 THC 合成提供了一种有吸引力的替代方案。我们目前正在研究这种策略在杂环合成中的进一步应用。
实验部分
转换 5-6 的一般程序
在 Ar 气氛下,向 5 (1 eq.) 的 CHCl3 (0.2 M) 搅拌溶液中加入三氟乙酸银 (1.25 eq.)。将混合物在 60 °C 下搅拌 30 分钟。通过过滤 Celite 垫除去沉淀的固体,并用 CH2Cl2 充分洗涤。混合滤液和洗涤液用 1 M NaOH 水溶液淬灭,用 H2O 稀释,然后用 CH2Cl2 萃取(3 次)。将合并的提取物干燥并在减压下浓缩。残留物在硅胶上通过柱层析纯化。
支持信息中给出了所有新化合物的详细程序和分析数据。
确认
这项研究得到了藤森科学技术基金会的支持。





























 京公网安备 11010802027423号
京公网安备 11010802027423号