当前位置:
X-MOL 学术
›
J. Phys. Chem. Lett.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Toward a Correct Description of Initial Electronic Coherence in Nonadiabatic Dynamics Simulations
The Journal of Physical Chemistry Letters ( IF 4.8 ) Pub Date : 2024-11-14 , DOI: 10.1021/acs.jpclett.4c02418 Jonathan R. Mannouch, Aaron Kelly
The Journal of Physical Chemistry Letters ( IF 4.8 ) Pub Date : 2024-11-14 , DOI: 10.1021/acs.jpclett.4c02418 Jonathan R. Mannouch, Aaron Kelly
|
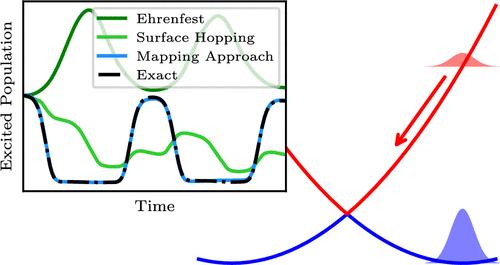
The recent improvement in experimental capabilities for interrogating and controlling molecular systems with ultrafast coherent light sources calls for the development of theoretical approaches that can accurately and efficiently treat electronic coherence. However, the most popular and practical nonadiabatic molecular dynamics techniques, Tully’s fewest-switches surface hopping and Ehrenfest mean-field dynamics, are unable to describe the dynamics proceeding from an initial electronic coherence. While such issues are not encountered with the analogous coupled-trajectory algorithms or numerically exact quantum dynamics methods, applying such techniques necessarily comes with a higher computational cost. Here we show that a correct description of initial electronic coherence can indeed be achieved using independent-trajectory methods derived from the semiclassical mapping formalism. The key is the introduction of an initial sampling over the electronic phase space and a means of incorporating phase interference between trajectories, both of which are naturally achieved when working within the semiclassical mapping framework.
中文翻译:

正确描述非绝热动力学仿真中的初始电子相干性
最近,使用超快相干光源询问和控制分子系统的实验能力的提高要求开发能够准确有效地处理电子相干性的理论方法。然而,最流行和最实用的非绝热分子动力学技术,Tully 的最少开关表面跳跃和 Ehrenfest 平均场动力学,无法描述从初始电子相干开始的动力学。虽然使用类似的耦合轨迹算法或数值精确的量子动力学方法不会遇到此类问题,但应用此类技术必然会带来更高的计算成本。在这里,我们表明,使用源自半经典映射形式主义的独立轨迹方法确实可以实现初始电子相干的正确描述。关键是在电子相空间上引入初始采样,以及一种在轨迹之间合并相位干扰的方法,这两者都是在半经典映射框架内工作时自然实现的。
更新日期:2024-11-14
中文翻译:
正确描述非绝热动力学仿真中的初始电子相干性
最近,使用超快相干光源询问和控制分子系统的实验能力的提高要求开发能够准确有效地处理电子相干性的理论方法。然而,最流行和最实用的非绝热分子动力学技术,Tully 的最少开关表面跳跃和 Ehrenfest 平均场动力学,无法描述从初始电子相干开始的动力学。虽然使用类似的耦合轨迹算法或数值精确的量子动力学方法不会遇到此类问题,但应用此类技术必然会带来更高的计算成本。在这里,我们表明,使用源自半经典映射形式主义的独立轨迹方法确实可以实现初始电子相干的正确描述。关键是在电子相空间上引入初始采样,以及一种在轨迹之间合并相位干扰的方法,这两者都是在半经典映射框架内工作时自然实现的。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号