npj Computational Materials ( IF 9.4 ) Pub Date : 2024-11-13 , DOI: 10.1038/s41524-024-01451-y Kazuma Ito, Tatsuya Yokoi, Katsutoshi Hyodo, Hideki Mori
|
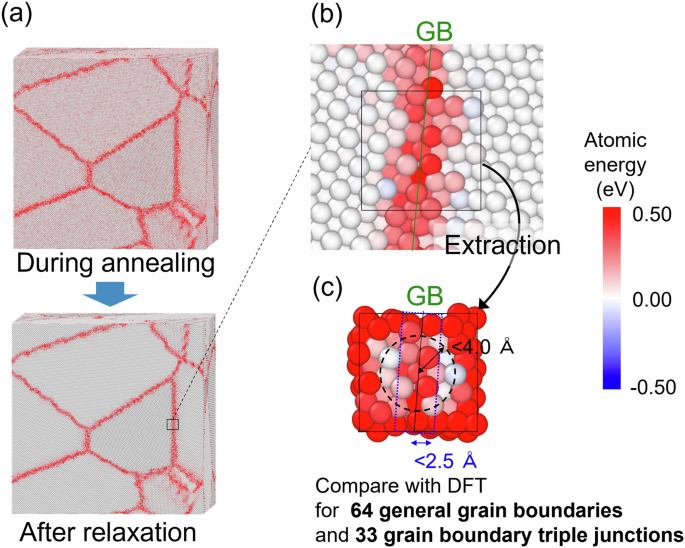
To advance the development of high-strength polycrystalline metallic materials towards achieving carbon neutrality, it is essential to design materials in which the atomic level control of general grain boundaries (GGBs), which govern the material properties, is achieved. However, owing to the complex and diverse structures of GGBs, there have been no reports on interatomic potentials capable of reproducing them. This accuracy is essential for conducting molecular dynamics analyses to derive material design guidelines. In this study, we constructed a machine learning interatomic potential (MLIP) with density functional theory (DFT) accuracy to model the energy, atomic structure, and dynamics of arbitrary grain boundaries (GBs), including GGBs, in α-Fe. Specifically, we employed a training dataset comprising diverse atomic structures generated based on crystal space groups. The GGB accuracy was evaluated by directly comparing with DFT calculations performed on cells cut near GBs from nano-polycrystals, and extrapolation grades of the local atomic environment based on active learning methods for the entire nano-polycrystal. Furthermore, we analyzed the GB energy and atomic structure in α-Fe polycrystals through large-scale molecular dynamics analysis using the constructed MLIP. The average GB energy of α-Fe polycrystals calculated by the constructed MLIP is 1.57 J/m2, exhibiting good agreement with experimental predictions. Our findings demonstrate the methodology for constructing an MLIP capable of representing GGBs with high accuracy, thereby paving the way for materials design based on computational materials science for polycrystalline materials.
中文翻译:
机器学习原子间势,对 Œ±-Fe 中一般晶界的 DFT 精度
为了推进高强度多晶金属材料的发展以实现碳中和,必须设计出能够实现控制材料特性的一般晶界 (GGB) 的原子级控制的材料。然而,由于 GGB 的结构复杂多样,目前还没有关于能够再现它们的原子间势的报道。这种准确性对于进行分子动力学分析以得出材料设计指南至关重要。在这项研究中,我们构建了一个具有密度泛函理论 (DFT) 精度的机器学习原子间势 (MLIP),以模拟 Œ±-Fe 中任意晶界 (GB) 的能量、原子结构和动力学,包括 GGB。具体来说,我们采用了一个训练数据集,其中包含基于晶体空间群生成的各种原子结构。通过直接比较对纳米多晶中在 GB 附近切割的单元进行的 DFT 计算,以及基于整个纳米多晶的主动学习方法的局部原子环境的外推等级,来评估 GGB 精度。此外,我们使用构建的 MLIP 通过大规模分子动力学分析分析了 Œ±-Fe 多晶中的 GB 能量和原子结构。通过构建的 MLIP 计算的 Œ±-Fe 多晶的平均 GB 能量为 1.57'ÄâJ/m2,与实验预测具有良好的一致性。我们的研究结果证明了构建能够高精度表示 GGB 的 MLIP 的方法,从而为基于多晶材料计算材料科学的材料设计铺平了道路。



















































 京公网安备 11010802027423号
京公网安备 11010802027423号