当前位置:
X-MOL 学术
›
J. Mater. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Structural and thermodynamic properties of the Li6PS5Cl solid electrolyte using first-principles calculations
Journal of Materials Chemistry A ( IF 10.7 ) Pub Date : 2024-11-11 , DOI: 10.1039/d4ta05159a Tarek Ayadi, Maylise Nastar, Fabien Bruneval
Journal of Materials Chemistry A ( IF 10.7 ) Pub Date : 2024-11-11 , DOI: 10.1039/d4ta05159a Tarek Ayadi, Maylise Nastar, Fabien Bruneval
|
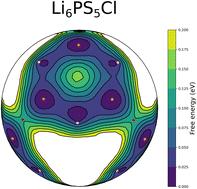
We perform static and dynamic ab initio simulations to investigate the structural and the thermodynamic properties of Li6PS5Cl, a solid electrolyte actively considered for solid-state batteries. Our simulations account for the disorder in the structure where the Li atoms can rotate either around sulfur or chlorine atoms. Li6PS5Cl presents a non-uniform distribution of Li ions around S and Cl atoms, which tends to become more homogeneous at higher temperature. This specific short-range order of Li has a significant impact on the stability of Li6PS5Cl. Comparing with recent X-ray and neutron diffraction studies, we confirm one Li crystallographic site position (Li1) and amend the coordinates of a second one (Li2). We then address the calculation of the heat capacity Cp with a combination of ab initio trajectories and a so-called temperature remapping approximation. Indeed, the standard quasi-harmonic approximation is not able to capture the complex energy landscape experienced by the mobile lithium atoms. To the best of our knowledge, there exists no experimental or theoretical Cp value for Li6PS5Cl in the literature, despite the importance of this thermodynamic quantity. Finally we use this more reliable Cp to investigate the thermodynamic stability of Li6PS5Cl against the decomposition reaction leading to Li2S, Li3PS4 and LiCl. We show that Li6PS5Cl is stable above 700 K, which is consistent with the high synthesis temperatures.
中文翻译:

使用第一性原理计算的 Li6PS5Cl 固体电解质的结构和热力学性质
我们执行静态和动态 ab initio 模拟,以研究 Li 6 PS 5 Cl 的结构和热力学特性,Li6PS5Cl 是一种积极考虑用于固态电池的固体电解质。我们的模拟解释了结构中的无序,其中 Li 原子可以围绕硫原子或氯原子旋转。Li6PS5Cl 在 S 和 Cl 原子周围呈现出不均匀的锂离子分布,在较高温度下趋于更加均匀。Li的这种特定的短程有序对Li6PS5Cl的稳定性有重大影响。与最近的X射线和中子衍射研究相比,我们确认了一个Li晶体位点位置(Li1)并修正了第二个位置(Li2)的坐标。然后,我们结合从头计算轨迹和所谓的温度重映射近似值来计算热容 C。事实上,标准的准谐波近似无法捕捉移动锂原子所经历的复杂能量景观。据我们所知,尽管这个热力学量很重要,但文献中没有 Li6PS5Cl 的实验或理论 Cp 值。 最后,我们利用这种更可靠的 Cp 来研究 Li6PS5Cl 对导致 Li2S、Li3PS4 和 LiCl 的分解反应的热力学稳定性。结果表明,Li6PS5Cl 在 700 K 以上是稳定的,这与高合成温度一致。
更新日期:2024-11-11
中文翻译:
使用第一性原理计算的 Li6PS5Cl 固体电解质的结构和热力学性质
我们执行静态和动态 ab initio 模拟,以研究 Li 6 PS 5 Cl 的结构和热力学特性,Li6PS5Cl 是一种积极考虑用于固态电池的固体电解质。我们的模拟解释了结构中的无序,其中 Li 原子可以围绕硫原子或氯原子旋转。Li6PS5Cl 在 S 和 Cl 原子周围呈现出不均匀的锂离子分布,在较高温度下趋于更加均匀。Li的这种特定的短程有序对Li6PS5Cl的稳定性有重大影响。与最近的X射线和中子衍射研究相比,我们确认了一个Li晶体位点位置(Li1)并修正了第二个位置(Li2)的坐标。然后,我们结合从头计算轨迹和所谓的温度重映射近似值来计算热容 C。事实上,标准的准谐波近似无法捕捉移动锂原子所经历的复杂能量景观。据我们所知,尽管这个热力学量很重要,但文献中没有 Li6PS5Cl 的实验或理论 Cp 值。 最后,我们利用这种更可靠的 Cp 来研究 Li6PS5Cl 对导致 Li2S、Li3PS4 和 LiCl 的分解反应的热力学稳定性。结果表明,Li6PS5Cl 在 700 K 以上是稳定的,这与高合成温度一致。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号