当前位置:
X-MOL 学术
›
Eur. J. Med. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Multicomponent syntheses enable the discovery of novel quisinostat-derived chemotypes as histone deacetylase inhibitors
European Journal of Medicinal Chemistry ( IF 6.0 ) Pub Date : 2024-11-08 , DOI: 10.1016/j.ejmech.2024.117045 Daniel Stopper, Susanna Buntrock, Kathrin Tan, Lais Pessanha de Carvalho, Linda Schäker-Hübner, Jana Held, Matthias U. Kassack, Finn K. Hansen
European Journal of Medicinal Chemistry ( IF 6.0 ) Pub Date : 2024-11-08 , DOI: 10.1016/j.ejmech.2024.117045 Daniel Stopper, Susanna Buntrock, Kathrin Tan, Lais Pessanha de Carvalho, Linda Schäker-Hübner, Jana Held, Matthias U. Kassack, Finn K. Hansen
|
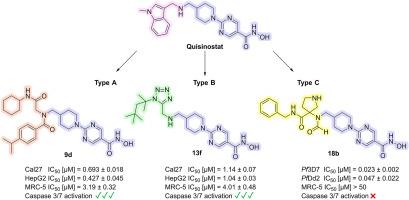
In this study, we synthesized and evaluated novel histone deacetylase (HDAC) inhibitors derived from the clinical candidate quisinostat. A library of 16 compounds categorized in three novel chemotypes was rapidly generated using multicomponent reactions (MCRs), enabling efficient structure-activity relationship studies. First, the compounds were evaluated for their activity against the Plasmodium falciparum strains 3D7 and Dd2, the main malaria-causing parasite, identifying compound 18b of the type C series as the most potent. It demonstrated low nanomolar IC50 values (IC50 (3D7) = 0.023 μM; IC50 (Dd2) = 0.047 μM) and high parasite selectivity (SIMRC−5/Pf 3D7 > 2174). HDAC inhibition assays confirmed substantial inhibition of the P. falciparum enzyme Pf HDAC1 (IC50 = 0.037 μM) as well as of human HDAC1 (IC50 = 0.021 μM) and HDAC6 (IC50 = 0.25 μM). Docking studies suggested distinct binding modes of 18b in P. falciparum and human HDAC1. Additionally, the in vitro anticancer activity was evaluated in Cal27 (head-neck carcinoma), HepG2 (hepatocellular carcinoma), A2780 (ovarian carcinoma), and U87 (glioblastoma) cell lines. Compounds 9b , 9d , and 13f showed potent antiproliferative activity and caspase 3/7 activation, in contrast to 18b . Furthermore, these compounds caused hyperacetylation of histone H3 and α-tubulin, indicating robust cellular target engagement. Overall, in this work we have identified the HDAC inhibitor 18b with selective antiplasmodial and 9b , 9d , and 13f with selective anticancer activities, providing valuable hits for further drug development efforts aimed at creating derivatives with reduced cytotoxicity against non-cancer cells compared to quisinostat.
中文翻译:

多组分合成能够发现新的呜呜虫病衍生的化学型作为组蛋白脱乙酰酶抑制剂
在这项研究中,我们合成并评价了源自临床候选喹诺司他的新型组蛋白脱乙酰酶 (HDAC) 抑制剂。使用多组分反应 (MCR) 快速生成了分为三种新型化学型的 16 种化合物库,从而实现了高效的构效关系研究。首先,评估了这些化合物对恶性疟原虫菌株 3D7 和 Dd2(主要引起疟疾的寄生虫)的活性,确定 C 型系列的化合物 18b 为最有效的化合物。它显示低纳摩尔 IC50 值 (IC50 (3D7) = 0.023 μM;IC50 (Dd2) = 0.047 μM) 和高寄生虫选择性 (SIMRC-5/Pf3D7 > 2174)。HDAC 抑制试验证实对恶性疟原虫酶 PfHDAC1 (IC50 = 0.037 μM) 以及人 HDAC1 (IC50 = 0.021 μM) 和 HDAC6 (IC50 = 0.25 μM) 具有显著抑制作用。对接研究表明 18b 在恶性疟原虫和人 HDAC1 中的结合模式不同。此外,在 Cal27 (头颈癌)、HepG2 (肝细胞癌)、A2780 (卵巢癌) 和 U87 (胶质母细胞瘤) 细胞系中评估了体外抗癌活性。与 18b 相比,化合物 9b、9d 和 13f 显示出强大的抗增殖活性和 caspase 3/7 激活。此外,这些化合物导致组蛋白 H3 和 α-微管蛋白的高乙酰化,表明细胞靶标具有很强的结合能力。总体而言,在这项工作中,我们已经确定了具有选择性抗疟药的 HDAC 抑制剂 18b 和具有选择性抗癌活性的 9b、9d 和 13f,为进一步的药物开发工作提供了有价值的命中,旨在创造与呜呜司他相比对非癌细胞具有降低细胞毒性的衍生物。
更新日期:2024-11-08
中文翻译:
多组分合成能够发现新的呜呜虫病衍生的化学型作为组蛋白脱乙酰酶抑制剂
在这项研究中,我们合成并评价了源自临床候选喹诺司他的新型组蛋白脱乙酰酶 (HDAC) 抑制剂。使用多组分反应 (MCR) 快速生成了分为三种新型化学型的 16 种化合物库,从而实现了高效的构效关系研究。首先,评估了这些化合物对恶性疟原虫菌株 3D7 和 Dd2(主要引起疟疾的寄生虫)的活性,确定 C 型系列的化合物 18b 为最有效的化合物。它显示低纳摩尔 IC50 值 (IC50 (3D7) = 0.023 μM;IC50 (Dd2) = 0.047 μM) 和高寄生虫选择性 (SIMRC-5/Pf3D7 > 2174)。HDAC 抑制试验证实对恶性疟原虫酶 PfHDAC1 (IC50 = 0.037 μM) 以及人 HDAC1 (IC50 = 0.021 μM) 和 HDAC6 (IC50 = 0.25 μM) 具有显著抑制作用。对接研究表明 18b 在恶性疟原虫和人 HDAC1 中的结合模式不同。此外,在 Cal27 (头颈癌)、HepG2 (肝细胞癌)、A2780 (卵巢癌) 和 U87 (胶质母细胞瘤) 细胞系中评估了体外抗癌活性。与 18b 相比,化合物 9b、9d 和 13f 显示出强大的抗增殖活性和 caspase 3/7 激活。此外,这些化合物导致组蛋白 H3 和 α-微管蛋白的高乙酰化,表明细胞靶标具有很强的结合能力。总体而言,在这项工作中,我们已经确定了具有选择性抗疟药的 HDAC 抑制剂 18b 和具有选择性抗癌活性的 9b、9d 和 13f,为进一步的药物开发工作提供了有价值的命中,旨在创造与呜呜司他相比对非癌细胞具有降低细胞毒性的衍生物。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号