当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Enhanced and Efficient Predictions of Dynamic Ionization through Constant-pH Adiabatic Free Energy Dynamics
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-11-08 , DOI: 10.1021/acs.jctc.4c00704 Richard S. Hong, Busayo D. Alagbe, Alessandra Mattei, Ahmad Y. Sheikh, Mark E. Tuckerman
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-11-08 , DOI: 10.1021/acs.jctc.4c00704 Richard S. Hong, Busayo D. Alagbe, Alessandra Mattei, Ahmad Y. Sheikh, Mark E. Tuckerman
|
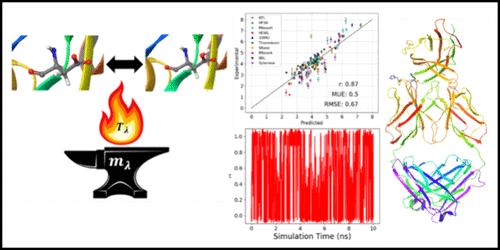
Dynamic or structurally induced ionization is a critical aspect of many physical, chemical, and biological processes. Molecular dynamics (MD) based simulation approaches, specifically constant pH MD methods, have been developed to simulate ionization states of molecules or proteins under experimentally or physiologically relevant conditions. While such approaches are now widely utilized to predict ionization sites of macromolecules or to study physical or biological phenomena, they are often computationally expensive and require long simulation times to converge. In this article, using the principles of adiabatic free energy dynamics, we introduce an efficient technique for performing constant pH MD simulations within the framework of the adiabatic free energy dynamics (AFED) approach. We call the new approach pH-AFED. We show that pH-AFED provides highly accurate predictions of protein residue pKa values, with a MUE of 0.5 pKa units when coupled with driven adiabatic free energy dynamics (d-AFED), while reducing the required simulation times by more than an order of magnitude. In addition, pH-AFED can be easily integrated into most constant pH MD codes or implementations and flexibly adapted to work in conjunction with enhanced sampling algorithms that target collective variables. We demonstrate that our approaches, with both pH-AFED standalone as well as pH-AFED combined with collective variable based enhanced sampling, provide promising predictive accuracy, with a MUE of 0.6 and 0.5 pKa units respectively, on a diverse range of proteins and enzymes, ranging up to 186 residues and 21 titratable sites. Lastly, we demonstrate how this approach can be utilized to understand the in vivo performance engineered antibodies for immunotherapy.
中文翻译:

通过恒定 pH 绝热自由能动力学增强和高效地预测动态电离
动态或结构诱导电离是许多物理、化学和生物过程的关键方面。基于分子动力学 (MD) 的模拟方法,特别是恒定 pH MD 方法,已被开发用于模拟分子或蛋白质在实验或生理相关条件下的电离状态。虽然这些方法现在被广泛用于预测大分子的电离位点或研究物理或生物现象,但它们的计算成本通常很高,并且需要较长的仿真时间才能收敛。在本文中,利用绝热自由能动力学的原理,我们介绍了一种在绝热自由能动力学 (AFED) 方法框架内执行恒定 pH MD 模拟的有效技术。我们将新方法称为 pH-AFED。我们表明,pH-AFED 提供了对蛋白质残基 pKa 值的高精度预测,当与驱动绝热自由能动力学 (d-AFED) 耦合时,MUE 为 0.5 pKa 单位,同时将所需的模拟时间缩短了一个数量级以上。此外,pH-AFED 可以轻松集成到大多数恒定 pH MD 代码或实施中,并灵活适应以与针对集体变量的增强采样算法结合使用。我们证明,我们的方法,包括 pH-AFED 独立以及 pH-AFED 与基于集体变量的增强采样相结合,提供了有希望的预测准确性,MUE 分别为 0.6 和 0.5 pK一个单位,适用于各种蛋白质和酶,范围高达 186 个残基和 21 个可滴定位点。 最后,我们展示了如何利用这种方法来了解用于免疫治疗的体内性能工程抗体。
更新日期:2024-11-08
中文翻译:
通过恒定 pH 绝热自由能动力学增强和高效地预测动态电离
动态或结构诱导电离是许多物理、化学和生物过程的关键方面。基于分子动力学 (MD) 的模拟方法,特别是恒定 pH MD 方法,已被开发用于模拟分子或蛋白质在实验或生理相关条件下的电离状态。虽然这些方法现在被广泛用于预测大分子的电离位点或研究物理或生物现象,但它们的计算成本通常很高,并且需要较长的仿真时间才能收敛。在本文中,利用绝热自由能动力学的原理,我们介绍了一种在绝热自由能动力学 (AFED) 方法框架内执行恒定 pH MD 模拟的有效技术。我们将新方法称为 pH-AFED。我们表明,pH-AFED 提供了对蛋白质残基 pKa 值的高精度预测,当与驱动绝热自由能动力学 (d-AFED) 耦合时,MUE 为 0.5 pKa 单位,同时将所需的模拟时间缩短了一个数量级以上。此外,pH-AFED 可以轻松集成到大多数恒定 pH MD 代码或实施中,并灵活适应以与针对集体变量的增强采样算法结合使用。我们证明,我们的方法,包括 pH-AFED 独立以及 pH-AFED 与基于集体变量的增强采样相结合,提供了有希望的预测准确性,MUE 分别为 0.6 和 0.5 pK一个单位,适用于各种蛋白质和酶,范围高达 186 个残基和 21 个可滴定位点。 最后,我们展示了如何利用这种方法来了解用于免疫治疗的体内性能工程抗体。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号