当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
DSDPFlex: Flexible-Receptor Docking with GPU Acceleration
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2024-11-08 , DOI: 10.1021/acs.jcim.4c01715 Chengwei Dong, Yu-Peng Huang, Xiaohan Lin, Hong Zhang, Yi Qin Gao
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2024-11-08 , DOI: 10.1021/acs.jcim.4c01715 Chengwei Dong, Yu-Peng Huang, Xiaohan Lin, Hong Zhang, Yi Qin Gao
|
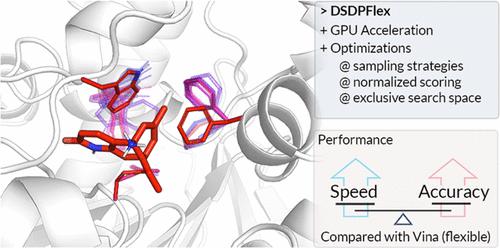
Molecular docking is an essential tool in structure-based drug discovery, widely utilized to model ligand–protein interactions and enrich potential hits. Among the different docking strategies, semiflexible docking (rigid-receptor and flexible-ligand model) is the most popular, benefiting from its balance of docking accuracy and speed. However, this approach ignores the conformational changes of proteins and hence demands suitable protein conformations as input. When the binding interaction adheres to an induced-fit model, flexible methods such as molecular dynamics simulation can be utilized, but they are computationally demanding. To balance between speed and accuracy, the flexible docking approach is an effective choice, as exemplified by AutoDock Vina and AutoDockFR, which treat selected protein side chains as flexible parts. However, the efficiency of flexible docking methods is yet to be improved for virtual screening usage. In this article, we introduce DSDPFlex, an improved flexible-receptor docking method accelerated by GPU parallelization. Beyond acceleration, optimizations with respect to sampling, scoring, and search space are implemented in DSDPFlex to further improve its capability in flexible tasks. In cross-docking evaluation, DSDPFlex demonstrates superior accuracy compared to AutoDock Vina and is 100 times faster than Vina in flexible-receptor tasks. We also show the advantage of flexible-receptor methods on suboptimal pockets and validate the advantage of DSDPFlex in screening on apo and AlphaFold2-predicted structures. With improvements in both efficiency and accuracy, DSDPFlex is expected to hold potential in future docking-based studies.
中文翻译:

DSDPFlex:具有 GPU 加速的灵活接收器对接
分子对接是基于结构的药物发现中的重要工具,广泛用于模拟配体-蛋白质相互作用和丰富潜在的命中。在不同的对接策略中,半柔性对接(刚性受体和柔性配体模型)是最受欢迎的,这得益于其对接精度和速度的平衡。然而,这种方法忽略了蛋白质的构象变化,因此需要合适的蛋白质构象作为输入。当结合相互作用遵循诱导拟合模型时,可以使用灵活的方法,例如分子动力学模拟,但它们对计算要求很高。为了在速度和准确性之间取得平衡,灵活的对接方法是一种有效的选择,AutoDock Vina 和 AutoDockFR 就是一个例子,它们将选定的蛋白质侧链视为柔性部分。然而,对于虚拟筛选的使用,灵活的对接方法的效率还有待提高。在本文中,我们介绍了 DSDPFlex,这是一种改进的柔性受体对接方法,由 GPU 并行化加速。除了加速之外,DSDPFlex 还实现了采样、评分和搜索空间方面的优化,以进一步提高其在灵活任务中的能力。在越库配送评估中,DSDPFlex 表现出优于 AutoDock Vina 的准确性,并且在柔性受体任务中比 Vina 快 100 倍。我们还展示了柔性受体方法对次优口袋的优势,并验证了 DSDPFlex 在筛选 apo 和 AlphaFold2 预测结构方面的优势。随着效率和准确性的提高,DSDPFlex 有望在未来基于对接的研究中具有潜力。
更新日期:2024-11-10
中文翻译:
DSDPFlex:具有 GPU 加速的灵活接收器对接
分子对接是基于结构的药物发现中的重要工具,广泛用于模拟配体-蛋白质相互作用和丰富潜在的命中。在不同的对接策略中,半柔性对接(刚性受体和柔性配体模型)是最受欢迎的,这得益于其对接精度和速度的平衡。然而,这种方法忽略了蛋白质的构象变化,因此需要合适的蛋白质构象作为输入。当结合相互作用遵循诱导拟合模型时,可以使用灵活的方法,例如分子动力学模拟,但它们对计算要求很高。为了在速度和准确性之间取得平衡,灵活的对接方法是一种有效的选择,AutoDock Vina 和 AutoDockFR 就是一个例子,它们将选定的蛋白质侧链视为柔性部分。然而,对于虚拟筛选的使用,灵活的对接方法的效率还有待提高。在本文中,我们介绍了 DSDPFlex,这是一种改进的柔性受体对接方法,由 GPU 并行化加速。除了加速之外,DSDPFlex 还实现了采样、评分和搜索空间方面的优化,以进一步提高其在灵活任务中的能力。在越库配送评估中,DSDPFlex 表现出优于 AutoDock Vina 的准确性,并且在柔性受体任务中比 Vina 快 100 倍。我们还展示了柔性受体方法对次优口袋的优势,并验证了 DSDPFlex 在筛选 apo 和 AlphaFold2 预测结构方面的优势。随着效率和准确性的提高,DSDPFlex 有望在未来基于对接的研究中具有潜力。
































 京公网安备 11010802027423号
京公网安备 11010802027423号