当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
QSLE-v1.0: A New Software Package for the Calculation of Coupled Quantum-Classical Dynamics in Condensed Phases Based on a Stochastic Approach
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-11-07 , DOI: 10.1021/acs.jctc.4c00960 Riccardo Cortivo, Mirco Zerbetto, Antonino Polimeno
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-11-07 , DOI: 10.1021/acs.jctc.4c00960 Riccardo Cortivo, Mirco Zerbetto, Antonino Polimeno
|
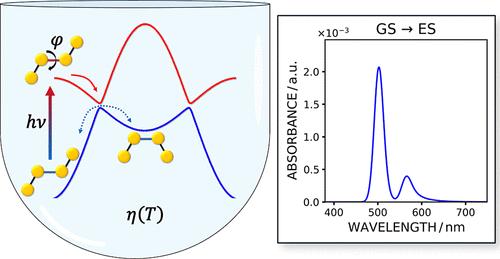
Recently, a stochastic version of the quantum-classical Liouville equation has been proposed [Campeggio, J.; Cortivo, R.; Zerbetto, M. J. Chem. Phys. 2023, 158, 244104], to compute the coupled quantum-classical dynamics of molecules in condensed phases. The approach, called quantum-stochastic Liouville equation (QSLE), is based on coupling the time evolution of electronic states to a stochastic description of the relevant (classical) nuclear coordinates. Natural internal coordinates are used, i.e., bond lengths, bond angles, and dihedral angles. The approach is tailored to directly propagate the populations of the electronic states over time, based on a classical Fokker–Planck equation for the nuclear degrees of freedom coupled to a master equation for the jumps among the electronic states. The QSLE is a multiscale approach requiring many ingredients to be assembled in order to carry out the numerical solution. To make the approach accessible, a software package that handles the main (and most cumbersome) details of the numerical workflow has been implemented into the software package QSLE-v1.0, which is introduced in the present paper. Here, it is considered the case of one torsional nuclear coordinate and two nonadiabatic electronic potential energy surfaces. This is a very basic model for interpreting photoisomerization or charge transfer phenomena, but despite its simplicity, it can be applied even in complex systems where the relevant quantum/classical parts affecting the phenomena under study are highly localized. A sand-box model system for describing photoisomerization processes is reported to demonstrate the usage of the software package. QSLE-v1.0 is open source and distributed under the GPLv2.0 license.
中文翻译:

QSLE-v1.0:基于随机方法计算凝聚相耦合量子-经典动力学的新软件包
最近,量子经典 Liouville 方程的随机版本被提出 [Campeggio, J.;科尔蒂沃,R.;Zerbetto, MJ 化学物理学。2023, 158, 244104],用于计算凝聚相中分子的耦合量子-经典动力学。这种方法称为量子随机 Liouville 方程 (QSLE),其基础是将电子状态的时间演变与相关(经典)核坐标的随机描述耦合。使用自然内部坐标,即键长、键角和二面角。该方法经过定制,可直接传播电子态的种群随时间变化,基于核自由度的经典 Fokker-Planck 方程与电子态之间跳跃的主方程耦合。QSLE 是一种多尺度方法,需要组装许多成分才能执行数值解。为了使该方法易于访问,本文介绍的软件包 QSLE-v1.0 中实现了一个处理数值工作流程的主要(也是最繁琐)细节的软件包。这里,它被认为是一个扭转核坐标和两个非绝热电子势能面的情况。这是解释光异构化或电荷转移现象的非常基本的模型,但尽管它很简单,但它甚至可以应用于影响所研究现象的相关量子/经典部分高度局部化的复杂系统。报道了用于描述光异构化过程的沙箱模型系统,以演示该软件包的用法。QSLE-v1.0 是开源的,并根据 GPLv2.0 许可证进行分发。
更新日期:2024-11-07
中文翻译:
QSLE-v1.0:基于随机方法计算凝聚相耦合量子-经典动力学的新软件包
最近,量子经典 Liouville 方程的随机版本被提出 [Campeggio, J.;科尔蒂沃,R.;Zerbetto, MJ 化学物理学。2023, 158, 244104],用于计算凝聚相中分子的耦合量子-经典动力学。这种方法称为量子随机 Liouville 方程 (QSLE),其基础是将电子状态的时间演变与相关(经典)核坐标的随机描述耦合。使用自然内部坐标,即键长、键角和二面角。该方法经过定制,可直接传播电子态的种群随时间变化,基于核自由度的经典 Fokker-Planck 方程与电子态之间跳跃的主方程耦合。QSLE 是一种多尺度方法,需要组装许多成分才能执行数值解。为了使该方法易于访问,本文介绍的软件包 QSLE-v1.0 中实现了一个处理数值工作流程的主要(也是最繁琐)细节的软件包。这里,它被认为是一个扭转核坐标和两个非绝热电子势能面的情况。这是解释光异构化或电荷转移现象的非常基本的模型,但尽管它很简单,但它甚至可以应用于影响所研究现象的相关量子/经典部分高度局部化的复杂系统。报道了用于描述光异构化过程的沙箱模型系统,以演示该软件包的用法。QSLE-v1.0 是开源的,并根据 GPLv2.0 许可证进行分发。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号