当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Hyperparameter Optimization for Atomic Cluster Expansion Potentials
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-11-06 , DOI: 10.1021/acs.jctc.4c01012 Daniel F. Thomas du Toit, Yuxing Zhou, Volker L. Deringer
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-11-06 , DOI: 10.1021/acs.jctc.4c01012 Daniel F. Thomas du Toit, Yuxing Zhou, Volker L. Deringer
|
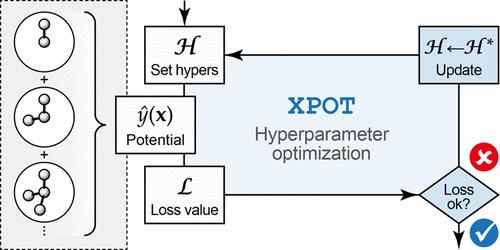
Machine learning-based interatomic potentials enable accurate materials simulations on extended time- and length scales. ML potentials based on the atomic cluster expansion (ACE) framework have recently shown promising performance for this purpose. Here, we describe a largely automated computational approach to optimizing hyperparameters for ACE potential models. We extend our openly available Python package, XPOT, to include an interface for ACE fitting, and discuss the optimization of the functional form and complexity of these models based on systematic sweeps across relevant hyperparameters. We showcase the usefulness of the approach for two example systems: the covalent network of silicon and the phase-change material Sb2Te3. More generally, our work emphasizes the importance of hyperparameter selection in the development of advanced ML potential models.
中文翻译:

原子簇扩展潜力的超参数优化
基于机器学习的原子间势能够在扩展的时间和长度尺度上进行准确的材料模拟。基于原子集群扩展 (ACE) 框架的 ML 潜力最近显示出可用于此目的的良好性能。在这里,我们描述了一种高度自动化的计算方法来优化 ACE 潜在模型的超参数。我们扩展了开放可用的 Python 包 XPOT,以包含用于 ACE 拟合的接口,并基于对相关超参数的系统扫描讨论了这些模型的功能形式和复杂性的优化。我们展示了该方法在两个示例系统中的有用性:硅的共价网络和相变材料 Sb2Te3。更一般地说,我们的工作强调了超参数选择在高级 ML 潜力模型开发中的重要性。
更新日期:2024-11-07
中文翻译:
原子簇扩展潜力的超参数优化
基于机器学习的原子间势能够在扩展的时间和长度尺度上进行准确的材料模拟。基于原子集群扩展 (ACE) 框架的 ML 潜力最近显示出可用于此目的的良好性能。在这里,我们描述了一种高度自动化的计算方法来优化 ACE 潜在模型的超参数。我们扩展了开放可用的 Python 包 XPOT,以包含用于 ACE 拟合的接口,并基于对相关超参数的系统扫描讨论了这些模型的功能形式和复杂性的优化。我们展示了该方法在两个示例系统中的有用性:硅的共价网络和相变材料 Sb2Te3。更一般地说,我们的工作强调了超参数选择在高级 ML 潜力模型开发中的重要性。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号