当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
SOP-MULTI: A Self-Organized Polymer-Based Coarse-Grained Model for Multidomain and Intrinsically Disordered Proteins with Conformation Ensemble Consistent with Experimental Scattering Data
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-11-05 , DOI: 10.1021/acs.jctc.4c00579 Krishnakanth Baratam, Anand Srivastava
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-11-05 , DOI: 10.1021/acs.jctc.4c00579 Krishnakanth Baratam, Anand Srivastava
|
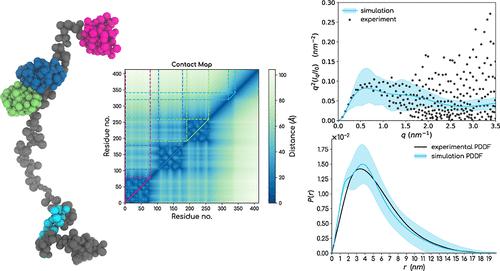
Multidomain proteins with long flexible linkers and full-length intrinsically disordered proteins (IDPs) are best defined as an ensemble of conformations rather than a single structure. Determining high-resolution ensemble structures of such proteins poses various challenges by using tools from experimental structural biophysics. Integrative approaches combining available low-resolution ensemble-averaged experimental data and in silico biomolecular reconstructions are now often used for the purpose. However, extensive Boltzmann weighted conformation sampling for large proteins, especially for ones where both the folded and disordered domains exist in the same polypeptide chain, remains a challenge. In this work, we present a 2-site per amino-acid resolution SOP-MULTI force field for simulating coarse-grained models of multidomain proteins. SOP-MULTI combines two well-established self-organized polymer models─: (i) SOP-SC models for folded systems and (ii) SOP-IDP for IDPs. For the SOP-MULTI, we introduce cross-interaction terms between the beads belonging to the folded and disordered regions to generate conformation ensembles for full-length multidomain proteins such as hnRNP A1, TDP-43, G3BP1, hGHR-ECD, TIA1, HIV-1 Gag, polyubiquitin, and FUS. When back-mapped to all-atom resolution, SOP-MULTI trajectories faithfully recapitulate the scattering data over the range of the reciprocal space. We also show that individual folded domains preserve native contacts with respect to solved folded structures, and root-mean-square fluctuations of residues in folded domains match those obtained from all-atom molecular dynamics simulation trajectories of the same folded systems. SOP-MULTI force field is made available as a LAMMPS-compatible user package along with setup codes for generating the required files for any full-length protein with folded and disordered regions.
中文翻译:

SOP-MULTI:一种基于自组织聚合物的粗粒模型,用于多结构域和内在无序蛋白质,其构象集成与实验散射数据一致
具有长柔性接头和全长固有无序蛋白 (IDP) 的多结构域蛋白最好定义为构象集合,而不是单一结构。通过使用实验结构生物物理学中的工具确定此类蛋白质的高分辨率集合结构带来了各种挑战。现在经常使用结合可用的低分辨率集成平均实验数据和计算机生物分子重建的综合方法来实现此目的。然而,对大蛋白进行广泛的玻尔兹曼加权构象采样,特别是对于折叠结构域和无序结构域都存在于同一多肽链中的蛋白质,仍然是一个挑战。在这项工作中,我们提出了一个每氨基酸分辨率 2 位点的 SOP-MULTI 力场,用于模拟多结构域蛋白质的粗粒模型。SOP-MULTI 结合了两个成熟的自组织聚合物模型:(i) 用于折叠系统的 SOP-SC 模型和 (ii) 用于 IDP 的 SOP-IDP。对于 SOP-MULTI,我们在属于折叠和无序区域的珠子之间引入交叉相互作用项,以生成全长多结构域蛋白的构象集合,例如 hnRNP A1、TDP-43、G3BP1、hGHR-ECD、TIA1、HIV-1 Gag、聚泛素和 FUS。当反向映射到全原子分辨率时,SOP-MULTI 轨迹忠实地概括了倒易空间范围内的散射数据。我们还表明,单个折叠结构域保留了相对于已解决的折叠结构的天然接触,并且折叠结构域中残基的均方根波动与从相同折叠系统的全原子分子动力学模拟轨迹中获得的波动相匹配。 SOP-MULTI 力场作为与 LAMMPS 兼容的用户包以及设置代码提供,用于为任何具有折叠和无序区域的全长蛋白质生成所需的文件。
更新日期:2024-11-06
中文翻译:
SOP-MULTI:一种基于自组织聚合物的粗粒模型,用于多结构域和内在无序蛋白质,其构象集成与实验散射数据一致
具有长柔性接头和全长固有无序蛋白 (IDP) 的多结构域蛋白最好定义为构象集合,而不是单一结构。通过使用实验结构生物物理学中的工具确定此类蛋白质的高分辨率集合结构带来了各种挑战。现在经常使用结合可用的低分辨率集成平均实验数据和计算机生物分子重建的综合方法来实现此目的。然而,对大蛋白进行广泛的玻尔兹曼加权构象采样,特别是对于折叠结构域和无序结构域都存在于同一多肽链中的蛋白质,仍然是一个挑战。在这项工作中,我们提出了一个每氨基酸分辨率 2 位点的 SOP-MULTI 力场,用于模拟多结构域蛋白质的粗粒模型。SOP-MULTI 结合了两个成熟的自组织聚合物模型:(i) 用于折叠系统的 SOP-SC 模型和 (ii) 用于 IDP 的 SOP-IDP。对于 SOP-MULTI,我们在属于折叠和无序区域的珠子之间引入交叉相互作用项,以生成全长多结构域蛋白的构象集合,例如 hnRNP A1、TDP-43、G3BP1、hGHR-ECD、TIA1、HIV-1 Gag、聚泛素和 FUS。当反向映射到全原子分辨率时,SOP-MULTI 轨迹忠实地概括了倒易空间范围内的散射数据。我们还表明,单个折叠结构域保留了相对于已解决的折叠结构的天然接触,并且折叠结构域中残基的均方根波动与从相同折叠系统的全原子分子动力学模拟轨迹中获得的波动相匹配。 SOP-MULTI 力场作为与 LAMMPS 兼容的用户包以及设置代码提供,用于为任何具有折叠和无序区域的全长蛋白质生成所需的文件。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号