当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
A Bond-Based Machine Learning Model for Molecular Polarizabilities and A Priori Raman Spectra
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-11-05 , DOI: 10.1021/acs.jctc.4c01086 Jakub K. Sowa, Peter J. Rossky
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-11-05 , DOI: 10.1021/acs.jctc.4c01086 Jakub K. Sowa, Peter J. Rossky
|
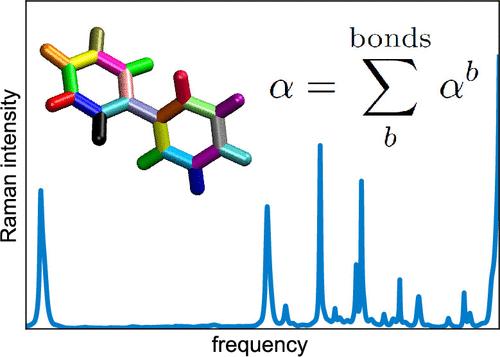
The use of machine learning (ML) algorithms in molecular simulations has become commonplace in recent years. There now exists, for instance, a multitude of ML force field algorithms that have enabled simulations approaching ab initio level accuracy at time scales and system sizes that significantly exceed what is otherwise possible with traditional methods. Far fewer algorithms exist for predicting rotationally equivariant, tensorial properties such as the electric polarizability. Here, we introduce a kernel ridge regression algorithm for machine learning of the polarizability tensor. This algorithm is based on the bond polarizability model and allows prediction of the tensor components at the cost similar to that of scalar quantities. We subsequently show the utility of this algorithm by simulating gas phase Raman spectra of biphenyl and malonaldehyde using classical molecular dynamics simulations of these systems performed with the recently developed MACE-OFF23 potential. The calculated spectra are shown to agree very well with the experiments and thus confirm the expediency of our algorithm as well as the accuracy of the used force field. More generally, this work demonstrates the potential of physics-informed approaches to yield simple yet effective machine learning algorithms for molecular properties.
中文翻译:

基于键的分子极化率和先验拉曼光谱机器学习模型
近年来,机器学习 (ML) 算法在分子模拟中的使用已变得司空见惯。例如,现在存在多种 ML 力场算法,这些算法使模拟能够在时间尺度和系统规模上接近从头到尾的精度,这大大超过了传统方法所能达到的水平。用于预测旋转等变张量特性(例如电极化率)的算法要少得多。在这里,我们介绍了一种核脊回归算法,用于极化率张量的机器学习。该算法基于键极化率模型,允许以类似于标量的成本预测张量分量。随后,我们通过使用最近开发的 MACE-OFF23 电位对这些系统进行经典分子动力学模拟来模拟联苯和丙二醛的气相拉曼光谱,从而展示了该算法的实用性。计算出的光谱与实验非常吻合,从而证实了我们算法的便利性以及所用力场的准确性。更一般地说,这项工作展示了以物理学为依据的方法在产生简单而有效的分子特性机器学习算法方面的潜力。
更新日期:2024-11-06
中文翻译:
基于键的分子极化率和先验拉曼光谱机器学习模型
近年来,机器学习 (ML) 算法在分子模拟中的使用已变得司空见惯。例如,现在存在多种 ML 力场算法,这些算法使模拟能够在时间尺度和系统规模上接近从头到尾的精度,这大大超过了传统方法所能达到的水平。用于预测旋转等变张量特性(例如电极化率)的算法要少得多。在这里,我们介绍了一种核脊回归算法,用于极化率张量的机器学习。该算法基于键极化率模型,允许以类似于标量的成本预测张量分量。随后,我们通过使用最近开发的 MACE-OFF23 电位对这些系统进行经典分子动力学模拟来模拟联苯和丙二醛的气相拉曼光谱,从而展示了该算法的实用性。计算出的光谱与实验非常吻合,从而证实了我们算法的便利性以及所用力场的准确性。更一般地说,这项工作展示了以物理学为依据的方法在产生简单而有效的分子特性机器学习算法方面的潜力。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号