当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
A Multipole-Based Reactive Force Field for Hydrocarbons
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-11-04 , DOI: 10.1021/acs.jctc.4c01285 Junben Weng, Hongqiang Cui, Da Zheng, Zhenhao Zhou, Dinglin Zhang, Huiying Chu, Anhui Wang, Guohui Li
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-11-04 , DOI: 10.1021/acs.jctc.4c01285 Junben Weng, Hongqiang Cui, Da Zheng, Zhenhao Zhou, Dinglin Zhang, Huiying Chu, Anhui Wang, Guohui Li
|
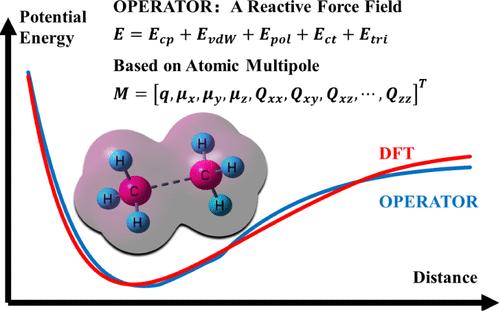
The computational complexity of quantum chemistry methods has prompted the development of reactive force fields, facilitating practical applications of molecular dynamics simulations for large-scale reactive systems. Current reactive force fields typically employ intricate corrections based on prior chemical knowledge, which severely impedes their further advancement. This study presents a new atomic multipole-based reactive model with bond free (OPERATOR). The force field is constructed on a simple, physically motivated model within the AMOEBA framework that closely resembles the physical representation of the chemical reaction processes. In the force field, the atomic multipoles are generated dynamically according to the atomic environments, aiming to effectively capture significant changes in the electrostatic environments during chemical reactions. Subsequently, atomic multipole-based charge penetration, polarization, and charge transfer effects are incorporated into the force field to describe the complex electrostatic interactions in the system. The force field also includes van der Waals interactions and three-body potentials. In addition, to extend these nonreactive interactions to chemical reactions, the atom distribution multipole moments are used to characterize different chemical environments. The force field has been optimized using the dataset of potential energy surfaces (PESs) of hydrocarbons derived from DFT results of millions of conformations with six degrees of freedom (DOFs). The results demonstrate that the new force field effectively replicates both the monopoles and the energies. In comparison to ReaxFF, the new force field exhibits comparable or superior performance. Furthermore, molecular dynamics simulations of n-heptane decomposition effectively reproduce the primary products and reactions observed in the experiments. Given the simplicity and physically motivated nature of the model, it is expected that the new force field will be utilized in future studies to investigate chemical reaction mechanisms involving more elements.
中文翻译:

碳氢化合物的基于多极子的反作用力场
量子化学方法的计算复杂性促进了反作用力场的发展,促进了分子动力学模拟在大规模反应系统的实际应用。当前的反作用力场通常采用基于先前化学知识的复杂校正,这严重阻碍了它们的进一步发展。本研究提出了一种新的基于原子多极子的反应模型,具有无键 (OPERATOR)。力场是在 AMOEBA 框架内一个简单的物理激励模型上构建的,该模型与化学反应过程的物理表示非常相似。在力场中,原子多极子是根据原子环境动态产生的,旨在有效捕捉化学反应过程中静电环境的显着变化。随后,基于原子多极子的电荷穿透、极化和电荷转移效应被纳入力场,以描述系统中复杂的静电相互作用。力场还包括范德华相互作用和三体势。此外,为了将这些非反应性相互作用扩展到化学反应,原子分布多极矩用于表征不同的化学环境。力场已使用碳氢化合物势能表面 (PES) 数据集进行了优化,该数据集来自具有六个自由度 (DOF) 的数百万个构象的 DFT 结果。结果表明,新的力场有效地复制了单极子和能量。与 ReaxFF 相比,新的力场表现出相当或更好的性能。 此外,正庚烷分解的分子动力学模拟有效地再现了实验中观察到的初级产物和反应。鉴于该模型的简单性和物理动机性质,预计新的力场将用于未来的研究中,以研究涉及更多元素的化学反应机制。
更新日期:2024-11-06
中文翻译:
碳氢化合物的基于多极子的反作用力场
量子化学方法的计算复杂性促进了反作用力场的发展,促进了分子动力学模拟在大规模反应系统的实际应用。当前的反作用力场通常采用基于先前化学知识的复杂校正,这严重阻碍了它们的进一步发展。本研究提出了一种新的基于原子多极子的反应模型,具有无键 (OPERATOR)。力场是在 AMOEBA 框架内一个简单的物理激励模型上构建的,该模型与化学反应过程的物理表示非常相似。在力场中,原子多极子是根据原子环境动态产生的,旨在有效捕捉化学反应过程中静电环境的显着变化。随后,基于原子多极子的电荷穿透、极化和电荷转移效应被纳入力场,以描述系统中复杂的静电相互作用。力场还包括范德华相互作用和三体势。此外,为了将这些非反应性相互作用扩展到化学反应,原子分布多极矩用于表征不同的化学环境。力场已使用碳氢化合物势能表面 (PES) 数据集进行了优化,该数据集来自具有六个自由度 (DOF) 的数百万个构象的 DFT 结果。结果表明,新的力场有效地复制了单极子和能量。与 ReaxFF 相比,新的力场表现出相当或更好的性能。 此外,正庚烷分解的分子动力学模拟有效地再现了实验中观察到的初级产物和反应。鉴于该模型的简单性和物理动机性质,预计新的力场将用于未来的研究中,以研究涉及更多元素的化学反应机制。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号