Advanced Synthesis & Catalysis ( IF 4.4 ) Pub Date : 2024-11-04 , DOI: 10.1002/adsc.202401251 Luca Nicchio, Lorenzo Di Terlizzi, Maurizio Fagnoni, Luc Neuville, Stefano Protti, Geraldine Masson
|
Introduction
The structural motif of 1,2,4-triazoles garners considerable attention due to its wide array of biological activities.1 These compounds have been extensively explored as selective enzyme inhibitors2-5 (including acetylcholinesterase, that plays a key role in the Alzheimer's Disease4), antimicrobials,1, 3 antivirals,6 anti-seizure,7 antifungals,8 analgesic,9 anticancer10 and agrochemicals11 (some significant examples are reported in Figure 1). Beyond pharmaceutical applications, electron-deficient 1,2,4-triazoles also exhibit excellent electron-transport and hole-blocking properties and have been extensively employed as ligands in transition-metal complexes and metal-organic frameworks (MOFs).12 Furthermore, this core has been successfully incorporated in polymeric materials for the fabrication of light emitting devices (e. g. OLEDs),13 photovoltaic cells,12 conductive membranes14 and even heat resistant explosives.15 Moreover, 3,5- or 1,5-disubstituted 1,2,4-triazoles are interesting bioisosteres of cis-amide bonds in peptides.16

Bioactive molecules containing the 1,2,4-triazole core.
Therefore, the synthesis of 1,2,4-triazoles has attracted considerable interest, resulting in the development of various synthetic strategies.17 These approaches typically emphasize condensation, cycloaddition reactions, or oxidative cyclization.17 Key starting materials such as imidates, amidrazones, aryldiazonium salts, and hydrazones are commonly employed as nitrogen sources in these processes.17 Despite the progress, challenges persist such as harsh reaction conditions (e. g. high temperature),17n reagent employed in stoichiometric excess,17l limited precursor stability17 and unfriendly reagents (e. g. gaseous and rather toxic trifluoroacetonitrile)17m as well as regioisomeric issues related to the preparation of polysubstituted triazoles.18 Consequently, there is a continuous drive to develop complementary methods that overcome these drawbacks and fulfill criteria encompassing novelty, versatility, operational practicality, and alignment with green chemistry principles to meet environmental requirements.
In this context, recent works took advantage of visible light-mediated approaches, which have proven to be both mild and efficient for the synthesis of various aza-cycles,19 to devise novel access to 1,2,4-triazoles. As such, a visible light photo-catalysed rearrangement of 1,2,4-oxadiazoles bearing a hydrazine side chain was reported to furnish triazoles.20a, 20b Another study developed the synthesis of 3-trifluoromethyl-1,2,4-triazoles through the photocycloaddition of sydnones with trifluoroacetonitrile, generated in situ from trifluoroacetaldehyde O-(aryl)oxime, using 4-CzIPN as the photocatalyst (Scheme 1a).20c Additionally, acridinium-catalyzed [3+2] photocyclization of azodicarboxylates and 2H-azirines was also shown to deliver 1,2,4-triazolines, which can be subsequently converted to 1,2,4-triazoles under basic conditions (Scheme 1b).20d A complementary three-component approach described access to 1,2,4-triazole-3,5-diamines from phenylhydrazines, isothiocyanato-benzenes, and 1,1,3,3-tetramethylguanidine, employing rose Bengal as the photocatalyst (Scheme 1c).20f More recently, access to polysubstituted 1,2,4-triazol-3-amines involving visible light Eosin Y-catalysed condensation of N,N-disubstituted hydrazines with N-cyano-N-aryl-p-toluenesulfonamides was also disclosed (Scheme 1d).20g

Examples of visible light -mediated synthesis of 1,2,4-triazoles.
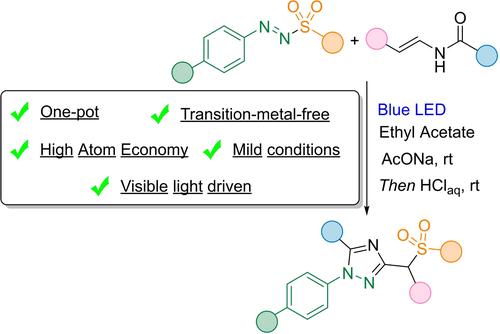
The potential of visible light in sustainable organic synthesis21 recently spurred our groups to design a regioselective and straightforward synthesis of α-sulfonyl arylhydrazones II via the direct 1,2-functionalization of styrenes I using photoactive bifunctional arylazo sulfones 122 and visible light (Scheme 2a).23 Due to the role of α-sulfonyl-hydrazones as precursors for the preparation of nitrogen-based heterocycles,20 we explored this approach for the direct synthesis of 1-aryl-1,2,4-triazoles. We hypothesized that upon irradiation, arylazo sulfones would generate sulfonyl and diazenyl radicals that could be directly and regioselectively incorporated into the C−C double bond of enamides leading to α-sulfonyl diazo derivatives IIIa, and the corresponding hydrazino-amides IIIb (Scheme 2b). Since such intermediates are known substrates of the named Pellizzari or Einhorn-Brunner triazole synthesis, IIIb should easily cyclize to form 1-aryl-3-sulfonylmethyl-1,2,4-triazoles III (Scheme 2c).24 Overall, such approach would be particularly attractive since it would benefit from cheap energetic source (visible light), would not need any stoichiometric or catalytic mediator and fully embed each part of the two starting materials in the target compounds by releasing only water.

a) Arylazo sulfones 1 for the visible-light double functionalization of styrenes; b) Our proposal; c) Einhorn-Brunner synthesis of 1,2,4-triazoles III.
中文翻译:
用芳氮砜法合成 1-芳基-3-磺酰甲基-1,2,4-三唑
介绍
1,2,4-三唑的结构基序因其广泛的生物活性而受到相当大的关注。1 这些化合物已被广泛探索为选择性酶抑制剂2-5(包括乙酰胆碱酯酶,它在阿尔茨海默病中起关键作用4)、抗菌剂 1,3 抗病毒药 6 抗癫痫药 7 抗真菌药 8 镇痛药 9 抗癌药10和农用化学品11(图 1 中报告了一些重要的例子)。除了制药应用外,缺电子的 1,2,4-三唑还表现出优异的电子传输和空穴阻挡特性,并已被广泛用作过渡金属络合物和金属有机框架 (MOF) 中的配体。12 此外,该核心已成功集成到聚合物材料中,用于制造发光器件(例如 OLED)、13 光伏电池、12 导电膜14 甚至耐热炸药。15 此外,3,5-或 1,5-二取代的 1,2,4-三唑是肽中顺酰胺键的有趣生物等排体。16
在图窗查看器PowerPoint 中打开
含有 1,2,4-三唑核心的生物活性分子。
因此,1,2,4-三唑类化合物的合成引起了相当大的兴趣,从而产生了各种合成策略。17 这些方法通常强调缩合、环加成反应或氧化环化。17 在这些过程中,亚胺酸盐、酰胺酮、芳基氮盐和腙等关键起始材料通常用作氮源。17 尽管取得了进展,但挑战仍然存在,例如苛刻的反应条件(例如高温)、17n 化学计量过量使用的试剂、17l 前驱体稳定性有限17 和不友好的试剂(例如气态和相当有毒的三氟乙腈)17m 以及与制备多取代三唑相关的区域异构体问题。18 因此,人们不断开发互补的方法,以克服这些缺点并满足包括新颖性、多功能性、操作实用性和符合绿色化学原则的标准,以满足环境要求。
在此背景下,最近的工作利用可见光介导的方法,这些方法已被证明对各种氮杂循环的合成既温和又有效,19 设计了对 1,2,4-三唑的新型访问。因此,据报道,带有肼侧链的 1,2,4-恶二唑的可见光光催化重排提供了三唑。20 个、20 个b另一项研究开发了通过使用 4-CzIPN 作为光催化剂,将三氟乙醛 O-(芳基)肟原位生成的三氟乙腈与三氟乙腈进行光环加成合成 3-三氟甲基-1,2,4-三唑(方案 1a)。20c 此外,吖啶催化的偶氮二羧酸盐和 2H-氮嗪的 [3+2] 光环化也显示可递送 1,2,4-三唑啉,随后在碱性条件下可转化为 1,2,4-三唑(方案 1b)。20d 一种互补的三组分方法描述了从苯肼、异硫氰基苯和 1,1,3,3-四甲基胍中获得 1,2,4-三唑-3,5-二胺,采用玫瑰孟加拉作为光催化剂(方案 1c)。20f 最近,还披露了涉及可见光曙红 Y 催化的 N,N-二取代肼与 N-氰基-N-芳基-对甲苯磺酰胺的缩合的多取代 1,2,4-三唑-3-胺(方案 1d)。20 克
在图窗查看器PowerPoint 中打开
可见光介导的 1,2,4-三唑合成实例。
可见光在可持续有机合成中的潜力 21 最近促使我们小组通过使用光活性双官能团芳氮砜 122 和可见光(方案 2a)对苯乙烯 I 进行直接 1,2-官能团化,设计了一种区域选择性和直接合成 α-磺酰芳基腙 II。23 由于 α-磺酰腙作为制备氮基杂环的前体的作用,20 我们探索了这种直接合成 1-芳基-1,2,4-三唑的方法。我们假设在照射时,芳氮砜会产生磺酰基和二氮酰基自由基,这些自由基可以直接和区域选择性地掺入烯酰胺的 C-C 双键中,导致 α-磺酰重氮衍生物 IIIa,以及相应的酰肼酰胺 IIIb(方案 2b)。由于此类中间体是命名的 Pellizzari 或 Einhorn-Brunner 三唑合成的已知底物,因此 IIIb 应该很容易环化形成 1-芳基-3-磺酰甲基-1,2,4-三唑 III(方案 2c)。24 总的来说,这种方法将特别有吸引力,因为它将受益于廉价的能量源(可见光),不需要任何化学计量或催化介质,并且只需释放水即可将两种起始材料的每个部分完全嵌入目标化合物中。
在图窗查看器PowerPoint 中打开
a) 芳氮砜 1 用于苯乙烯的可见光双官能团化;b) 我们的提案;c) 1,2,4-三唑类化合物 III 的 Einhorn-Brunner 合成。



















































 京公网安备 11010802027423号
京公网安备 11010802027423号