当前位置:
X-MOL 学术
›
Dalton Trans.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
A mechanistic study of chiral manganese porphyrin-catalyzed enantioselective C–H hydroxylation reaction
Dalton Transactions ( IF 3.5 ) Pub Date : 2024-11-04 , DOI: 10.1039/d4dt02452d Jing-Kun Gao, Wandong Chen, Junjie Tai, Zhengwei Chen, Hang Liu, Yuxin Du, Yiting Jiang, Yuanbin She, Yun-Fang Yang
Dalton Transactions ( IF 3.5 ) Pub Date : 2024-11-04 , DOI: 10.1039/d4dt02452d Jing-Kun Gao, Wandong Chen, Junjie Tai, Zhengwei Chen, Hang Liu, Yuxin Du, Yiting Jiang, Yuanbin She, Yun-Fang Yang
|
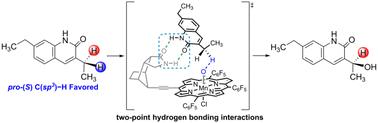
We employed density functional theory (DFT) calculations to elucidate the mechanism and origin of enantioselectivity in the C–H hydroxylation reaction catalyzed by a chiral manganese porphyrin complex. Our study reveals that the chiral manganese porphyrin forms a two-point hydrogen bonding interaction with the substrate. Specifically, the hydrogen atom abstraction of the methylene pro-(S) C–H bond at the heterocyclic C-3 position is 1.9 kcal mol−1 favored over the hydrogen atom abstraction of the pro-(R) C–H bond. This preferential reactivity results in the predominant formation of (S)-hydroxylated products. Our DFT calculations are consistent with the experimental findings of high enantioselectivity in the chiral manganese porphyrin catalyzed C(sp3)–H hydroxylation of lactam derivatives. The observed enantioselectivity arises from the formation of two-point hydrogen bonding between lactam derivatives and manganese porphyrin catalysts. Moreover, our computations indicate varying degrees of substrate distortion upon attack by high-valent manganese oxygen complexes at different hydrogen atoms.
中文翻译:

手性锰卟啉催化的对映选择性 C-H 羟基化反应的机理研究
我们采用密度泛函理论 (DFT) 计算来阐明手性锰卟啉复合物催化的 C-H 羟基化反应中对映选择性的机制和来源。我们的研究表明,手性锰卟啉与底物形成两点氢键相互作用。具体来说,亚甲基 pro-(S) C-H 键在杂环 C-3 位置的氢原子提取为 1.9 kcal mol-1,优于 pro-(R) C-H 键的氢原子提取。这种优先反应性导致 (S)-羟基化产物的主要形成。我们的 DFT 计算与手性锰卟啉催化的内酰胺衍生物 C(sp3)-H 羟基化反应中高对映选择性的实验结果一致。观察到的对映选择性源于内酰胺衍生物和锰卟啉催化剂之间形成的两点氢键。此外,我们的计算表明,在不同氢原子处受到高价锰氧络合物的攻击时,不同程度的衬底变形。
更新日期:2024-11-04
中文翻译:
手性锰卟啉催化的对映选择性 C-H 羟基化反应的机理研究
我们采用密度泛函理论 (DFT) 计算来阐明手性锰卟啉复合物催化的 C-H 羟基化反应中对映选择性的机制和来源。我们的研究表明,手性锰卟啉与底物形成两点氢键相互作用。具体来说,亚甲基 pro-(S) C-H 键在杂环 C-3 位置的氢原子提取为 1.9 kcal mol-1,优于 pro-(R) C-H 键的氢原子提取。这种优先反应性导致 (S)-羟基化产物的主要形成。我们的 DFT 计算与手性锰卟啉催化的内酰胺衍生物 C(sp3)-H 羟基化反应中高对映选择性的实验结果一致。观察到的对映选择性源于内酰胺衍生物和锰卟啉催化剂之间形成的两点氢键。此外,我们的计算表明,在不同氢原子处受到高价锰氧络合物的攻击时,不同程度的衬底变形。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号