当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Correction to Ab Initio Vibro-Polaritonic Spectra in Strongly Coupled Cavity-Molecule Systems
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-10-31 , DOI: 10.1021/acs.jctc.4c01374 Thomas Schnappinger, Markus Kowalewski
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-10-31 , DOI: 10.1021/acs.jctc.4c01374 Thomas Schnappinger, Markus Kowalewski
|
Recently, we have discovered several errors in our paper “Ab Initio Vibro-Polaritonic Spectra in Strongly Coupled Cavity-Molecule Systems” [Schnappinger and Kowalewski J. Chem.
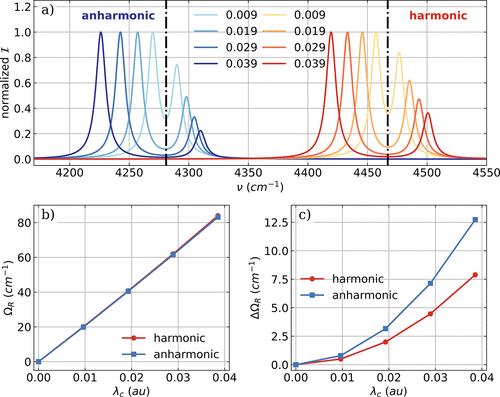
Theory Comput. 2023, 19, 9278] which we would like to correct here. The expression of the infrared (IR) intensity for the harmonic approximation given in eq 15 is lacking the atomic masses and should read instead Figure 1. a) Vibro-polaritonic IR spectra of a single HF molecule calculated in the harmonic approximation (reddish) and in the full-quantum setup (bluish), individually normalized for each coupling strength. The black dashed dotted lines indicate the bare molecular frequencies of the fundamental transitions of the harmonic case (Hν1 = 4467 cm–1) and the anharmonic case (Aν1 = 4281 cm–1). The cavity frequency ωc is resonant with the corresponding fundamental transition, and the coupling strength λc increases from 0.009 au to 0.039 au (from lightest to darkest color). The Rabi splitting ΩR (b) and its asymmetry ΔΩR =ωc – 0.5(νLP+νUP) (c) as a function of λc. Figure 4. a) Vibro-polaritonic IR spectra of a single HF molecule calculated in the harmonic approximation (reddish) and in the full-quantum setup (bluish), individually normalized for each coupling strength. In both cases, the full SCF treatment is neglected; for details, see text. Black dashed-dotted lines indicate the frequencies of the harmonic fundamental transition (Hν1 = 4467 cm–1) and the anharmonic fundamental transition (Aν1 = 4281 cm–1). The cavity frequency ωc is resonant with the corresponding fundamental transition in both cases, and the coupling strength λc increases from 0.009 au to 0.039 au (from light to dark color). b) Rabi splitting ΩR as a function of λc. c) Asymmetry ΔΩR = ωc – 0.5(νLP+νUP) of the Rabi splitting. Figure 5. Vibro-polaritonic IR spectra calculated in the harmonic approximation for different numbers of HF molecules (color-coded) shown with respect to the cavity frequency ωc. The cavity is resonant with the harmonic fundamental transition (Hν1 = 4467 cm–1, black dashed-dotted lines) and a rescaled coupling strength of λ0 of 0.057 au is used (see eq 17). a) Full CBO-HF simulation in the all-parallel configuration. b) Full CBO-HF simulation in the antiparallel configuration. c) Linear CBO-HF simulation (without DSE terms) in the all-parallel configuration. c) Linear CBO-HF simulation (without DSE terms) in the antiparallel configuration. Figure 7. a) Vibronic IR spectra of a single NH3 molecule calculated in the harmonic approximation. The low energy part of the vibro-polaritonic IR spectra of a single NH3 molecule zoomed into the symmetric mode (b,d) and the two asymmetric bending modes shown in (c,e). The polarization axis of the cavity mode is the y axis for (b,c) and equal to the z axis for (d,e). The cavity frequency ωc is resonant with the symmetric bending mode (1103 cm–1) and the cavity field strength λc increases from 0.039 au to 0.155 au. The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jctc.4c01374. Corrected Figures S3, S4, and S13 (PDF) Correction to Ab
Initio Vibro-Polaritonic Spectra
in Strongly Coupled Cavity-Molecule Systems 3 views 0 shares 0 downloads Most electronic Supporting Information files are available without a subscription to ACS Web Editions. Such files may be downloaded by article for research use (if there is a public use license linked to the relevant article, that license may permit other uses). Permission may be obtained from ACS for other uses through requests via the RightsLink permission system: http://pubs.acs.org/page/copyright/permissions.html. This article has not yet been cited by other publications.
中文翻译:

在强耦合腔-分子系统中对 Ab Initio Vibro-Polaritonic 光谱的校正
最近,我们在论文“强耦合腔-分子系统中的 Ab Initio Vibro-Polaritonic Spectra”中发现了几个错误 [Schnappinger 和 Kowalewski J. Chem. Theory Comput.2023, 19, 9278] 我们想在这里更正。红外 (IR) 强度的表达式
因为方程 15 中给出的谐波近似值缺少原子质量,应改为图 1。a) 在谐波近似(偏红)和全量子设置(偏蓝)中计算的单个 HF 分子的振极-偏振红外光谱,并针对每个耦合强度单独归一化。黑色虚线表示谐波情况 (Hν1 = 4467 cm–1) 和非谐波情况 (Aν1 = 4281 cm–1) 的基跃的裸分子频率。腔频率 ωc 与相应的基跃相谐振,耦合强度 λc 从 0.009 au 增加到 0.039 au(从最浅的颜色到最暗的颜色)。拉比分裂 ΩR (b) 及其不对称性 ΔΩR =ωc – 0.5(νLP+νUP) (c) 作为 λc 的函数。图 4.a) 在谐波近似(偏红)和全量子设置(偏蓝)中计算的单个 HF 分子的振极-偏振红外光谱,并针对每个耦合强度单独归一化。在这两种情况下,都忽略了完整的 SCF 治疗;有关详细信息,请参阅文本。黑色虚线表示谐波基频跃迁 (Hν1 = 4467 cm–1) 和非谐波基频跃迁 (Aν1 = 4281 cm–1) 的频率。在这两种情况下,腔频率 ωc 都与相应的基波跃迁谐振,耦合强度 λc 从 0.009 au 增加到 0.039 au(从浅色到深色)。 b) Rabi 分裂 ΩR 与 λc 的函数关系。c) 拉比分裂的不对称性 ΔΩR = ωc – 0.5(νLP+νUP)。图 5.振动偏振子红外光谱,以不同数量的 HF 分子(颜色编码)的谐波近似计算,显示相对于腔体频率 ωc。腔体与谐波基波跃迁(Hν1 = 4467 cm–1,黑色虚线)谐振,并使用 0.057 au 的 λ0 重新缩放的耦合强度(见方程 17)。a) 全并行配置中的全 CBO-HF 仿真。b) 反并行配置中的全 CBO-HF 仿真。c) 全并行配置中的线性 CBO-HF 仿真(无 DSE 项)。c) 反并行配置中的线性 CBO-HF 模拟(无 DSE 项)。图 7.a) 以谐波近似计算的单个 NH3 分子的振动红外光谱。单个 NH3 分子的振动偏振红外光谱的低能量部分放大到对称模式 (b,d) 和 (c,e) 中所示的两种不对称弯曲模式。腔模式的极化轴是 (b,c) 的 y 轴,等于 (d,e) 的 z 轴。腔频率 ωc 与对称弯曲模式 (1103 cm–1) 谐振,腔体场强 λc 从 0.039 au 增加到 0.155 au。支持信息可在 https://pubs.acs.org/doi/10.1021/acs.jctc.4c01374 免费获取。更正后的图 S3、S4 和 S13 (PDF) 对 Ab 的校正 Initio 振动-偏振子光谱 在 强耦合腔分子系统中 3 视图 0 分享 0 下载 大多数电子支持信息文件无需订阅 ACS Web Editions 即可获得。此类文件可以按文章下载用于研究用途(如果有链接到相关文章的公共使用许可证,则该许可证可能允许其他用途)。可以通过 RightsLink 权限系统(http://pubs.acs.org/page/copyright/permissions.html)请求,从 ACS 获得用于其他用途的权限。本文尚未被其他出版物引用。
更新日期:2024-11-02
中文翻译:
在强耦合腔-分子系统中对 Ab Initio Vibro-Polaritonic 光谱的校正
最近,我们在论文“强耦合腔-分子系统中的 Ab Initio Vibro-Polaritonic Spectra”中发现了几个错误 [Schnappinger 和 Kowalewski J. Chem. Theory Comput.2023, 19, 9278] 我们想在这里更正。红外 (IR) 强度的表达式
因为方程 15 中给出的谐波近似值缺少原子质量,应改为图 1。a) 在谐波近似(偏红)和全量子设置(偏蓝)中计算的单个 HF 分子的振极-偏振红外光谱,并针对每个耦合强度单独归一化。黑色虚线表示谐波情况 (Hν1 = 4467 cm–1) 和非谐波情况 (Aν1 = 4281 cm–1) 的基跃的裸分子频率。腔频率 ωc 与相应的基跃相谐振,耦合强度 λc 从 0.009 au 增加到 0.039 au(从最浅的颜色到最暗的颜色)。拉比分裂 ΩR (b) 及其不对称性 ΔΩR =ωc – 0.5(νLP+νUP) (c) 作为 λc 的函数。图 4.a) 在谐波近似(偏红)和全量子设置(偏蓝)中计算的单个 HF 分子的振极-偏振红外光谱,并针对每个耦合强度单独归一化。在这两种情况下,都忽略了完整的 SCF 治疗;有关详细信息,请参阅文本。黑色虚线表示谐波基频跃迁 (Hν1 = 4467 cm–1) 和非谐波基频跃迁 (Aν1 = 4281 cm–1) 的频率。在这两种情况下,腔频率 ωc 都与相应的基波跃迁谐振,耦合强度 λc 从 0.009 au 增加到 0.039 au(从浅色到深色)。 b) Rabi 分裂 ΩR 与 λc 的函数关系。c) 拉比分裂的不对称性 ΔΩR = ωc – 0.5(νLP+νUP)。图 5.振动偏振子红外光谱,以不同数量的 HF 分子(颜色编码)的谐波近似计算,显示相对于腔体频率 ωc。腔体与谐波基波跃迁(Hν1 = 4467 cm–1,黑色虚线)谐振,并使用 0.057 au 的 λ0 重新缩放的耦合强度(见方程 17)。a) 全并行配置中的全 CBO-HF 仿真。b) 反并行配置中的全 CBO-HF 仿真。c) 全并行配置中的线性 CBO-HF 仿真(无 DSE 项)。c) 反并行配置中的线性 CBO-HF 模拟(无 DSE 项)。图 7.a) 以谐波近似计算的单个 NH3 分子的振动红外光谱。单个 NH3 分子的振动偏振红外光谱的低能量部分放大到对称模式 (b,d) 和 (c,e) 中所示的两种不对称弯曲模式。腔模式的极化轴是 (b,c) 的 y 轴,等于 (d,e) 的 z 轴。腔频率 ωc 与对称弯曲模式 (1103 cm–1) 谐振,腔体场强 λc 从 0.039 au 增加到 0.155 au。支持信息可在 https://pubs.acs.org/doi/10.1021/acs.jctc.4c01374 免费获取。更正后的图 S3、S4 和 S13 (PDF) 对 Ab 的校正 Initio 振动-偏振子光谱 在 强耦合腔分子系统中 3 视图 0 分享 0 下载 大多数电子支持信息文件无需订阅 ACS Web Editions 即可获得。此类文件可以按文章下载用于研究用途(如果有链接到相关文章的公共使用许可证,则该许可证可能允许其他用途)。可以通过 RightsLink 权限系统(http://pubs.acs.org/page/copyright/permissions.html)请求,从 ACS 获得用于其他用途的权限。本文尚未被其他出版物引用。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号