当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Constructing Accurate and Efficient General-Purpose Atomistic Machine Learning Model with Transferable Accuracy for Quantum Chemistry
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-10-31 , DOI: 10.1021/acs.jctc.4c01151 Yicheng Chen, Wenjie Yan, Zhanfeng Wang, Jianming Wu, Xin Xu
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-10-31 , DOI: 10.1021/acs.jctc.4c01151 Yicheng Chen, Wenjie Yan, Zhanfeng Wang, Jianming Wu, Xin Xu
|
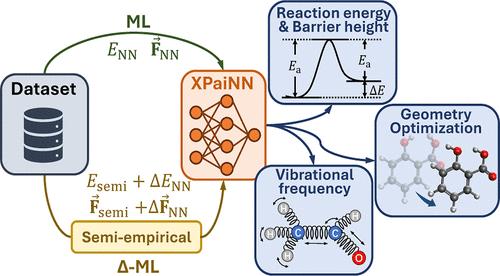
Density functional theory (DFT) has been a cornerstone in computational science, providing powerful insights into structure–property relationships for molecules and materials through first-principles quantum-mechanical (QM) calculations. However, the advent of atomistic machine learning (ML) is reshaping the landscape by enabling large-scale dynamics simulations and high-throughput screening at DFT-equivalent accuracy with drastically reduced computational cost. Yet, the development of general-purpose atomistic ML models as surrogates for QM calculations faces several challenges, particularly in terms of model capacity, data efficiency, and transferability across chemically diverse systems. This work introduces a novel extension of the polarizable atom interaction neural network (namely, XPaiNN) to address these challenges. Two distinct training strategies have been employed, one direct-learning and the other Δ-ML on top of a semiempirical QM method. These methodologies have been implemented within the same framework, allowing for a detailed comparison of their results. The XPaiNN models, in particular the one using Δ-ML, not only demonstrate competitive performance on standard benchmarks, but also demonstrate the effectiveness against other ML models and QM methods on comprehensive downstream tasks, including noncovalent interactions, reaction energetics, barrier heights, geometry optimization and reaction thermodynamics, etc. This work represents a significant step forward in the pursuit of accurate and efficient atomistic ML models of general-purpose, capable of handling complex chemical systems with transferable accuracy.
中文翻译:

为量子化学构建准确高效的具有可转移精度的通用原子机器学习模型
密度泛函理论 (DFT) 一直是计算科学的基石,它通过第一性原理量子力学 (QM) 计算为分子和材料的结构-性质关系提供了强大的见解。然而,原子机器学习 (ML) 的出现正在重塑格局,它能够以 DFT 等效的精度实现大规模动力学模拟和高通量筛选,同时大幅降低计算成本。然而,开发通用原子 ML 模型作为 QM 计算的替代物面临着一些挑战,特别是在模型容量、数据效率和跨化学不同系统的可转移性方面。这项工作引入了极化原子相互作用神经网络(即 XPaiNN)的新扩展来应对这些挑战。采用了两种不同的训练策略,一种是直接学习,另一种是半经验 QM 方法之上的 Δ-ML。这些方法已在同一框架内实施,允许对它们的结果进行详细比较。XPaiNN 模型,尤其是使用 Δ-ML 的模型,不仅在标准基准上表现出有竞争力的性能,而且还证明了在综合下游任务(包括非共价相互作用、反应能量学、势垒高度、几何优化和反应热力学等)上与其他 ML 模型和 QM 方法相比的有效性。这项工作代表了在追求准确高效的通用原子 ML 模型方面向前迈出的重要一步,能够以可转移的准确性处理复杂的化学系统。
更新日期:2024-10-31
中文翻译:
为量子化学构建准确高效的具有可转移精度的通用原子机器学习模型
密度泛函理论 (DFT) 一直是计算科学的基石,它通过第一性原理量子力学 (QM) 计算为分子和材料的结构-性质关系提供了强大的见解。然而,原子机器学习 (ML) 的出现正在重塑格局,它能够以 DFT 等效的精度实现大规模动力学模拟和高通量筛选,同时大幅降低计算成本。然而,开发通用原子 ML 模型作为 QM 计算的替代物面临着一些挑战,特别是在模型容量、数据效率和跨化学不同系统的可转移性方面。这项工作引入了极化原子相互作用神经网络(即 XPaiNN)的新扩展来应对这些挑战。采用了两种不同的训练策略,一种是直接学习,另一种是半经验 QM 方法之上的 Δ-ML。这些方法已在同一框架内实施,允许对它们的结果进行详细比较。XPaiNN 模型,尤其是使用 Δ-ML 的模型,不仅在标准基准上表现出有竞争力的性能,而且还证明了在综合下游任务(包括非共价相互作用、反应能量学、势垒高度、几何优化和反应热力学等)上与其他 ML 模型和 QM 方法相比的有效性。这项工作代表了在追求准确高效的通用原子 ML 模型方面向前迈出的重要一步,能够以可转移的准确性处理复杂的化学系统。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号