当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
MOFSynth: A Computational Tool toward Synthetic Likelihood Predictions of MOFs
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2024-10-31 , DOI: 10.1021/acs.jcim.4c01298 Charalampos G. Livas, Pantelis N. Trikalitis, George E. Froudakis
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2024-10-31 , DOI: 10.1021/acs.jcim.4c01298 Charalampos G. Livas, Pantelis N. Trikalitis, George E. Froudakis
|
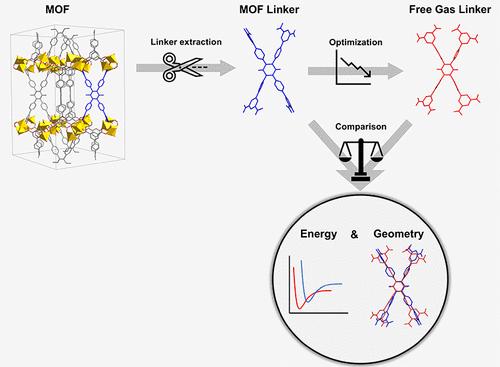
In the past decade, high-throughput computational studies of materials have increased significantly mainly due to advances in computer capabilities and have attracted a great deal of interest. In the field of metal–organic frameworks (MOFs), over a million hypothetical MOFs have been designed in silico, yet only a small fraction of these have been synthesized. For validating the computational-hypothetical results and accelerating the progress in the field, there is a pressing need for distinguishing MOFs that are more likely to be synthesized for real-life applications. This study presents a comprehensive investigation into the synthesizability likelihood of MOFs, utilizing a novel computational approach based on the disparities in energy and geometry between the linker conformation within the MOF structure and its isolated, free-gas state since both of these have been proven to be critical factors influencing MOF synthesis. Our user-friendly tool streamlines synthesizability evaluation, requiring minimal expertise in computational chemistry. By deconstructing over 40,000 MOFs from databases, including QMOF, CoRE MOF, and ToBaCCo, we analyze key parameters defining the linker strain within the MOF unit cell. Our results indicate that QMOF and CoRE MOF contain more promising candidates for synthesis, while ToBaCCo exhibits a relatively poor synthesizability likelihood due to unoptimized materials. Through extensive analysis, we identify optimal linker candidates for highly synthesizable MOFs. Consistent trends in energy distribution across databases that are confirmed by high Pearson and Spearman coefficients suggest the potential for omitting optimization calculations, significantly reducing computational costs. This study underscores the importance of linker deformation and energy disparities and enhances our understanding of synthetic accessibility in MOF research, offering valuable insights for future advancements in the field.
中文翻译:

MOFSynth:用于 MOF 合成似然预测的计算工具
在过去十年中,材料的高通量计算研究显着增加,这主要是由于计算机能力的进步,并引起了人们的极大兴趣。在金属有机框架 (MOF) 领域,已经用计算机设计了超过 100 万个假设的 MOF,但其中只有一小部分被合成。为了验证计算假设结果并加速该领域的进展,迫切需要区分更有可能合成用于实际应用的 MOF。本研究对 MOF 的可合成可能性进行了全面调查,利用一种新颖的计算方法,该方法基于 MOF 结构内的接头构象与其分离的自由气体状态之间的能量和几何形状之间的差异,因为这两者都已被证明是影响 MOF 合成的关键因素。我们的用户友好型工具简化了可合成性评估,只需极少的计算化学专业知识。通过从数据库(包括 QMOF、CoRE MOF 和 ToBaCCo)中解构超过 40,000 个 MOF,我们分析了定义 MOF 晶胞内接头菌株的关键参数。我们的结果表明,QMOF 和 CoRE MOF 包含更有前途的合成候选者,而 ToBaCCo 由于材料未优化,表现出相对较差的可合成性可能性。通过广泛的分析,我们确定了高度可合成 MOF 的最佳接头候选者。由高 Pearson 和 Spearman 系数证实的跨数据库能量分布的一致趋势表明,有可能省略优化计算,从而显著降低计算成本。 这项研究强调了接头变形和能量差异的重要性,并增强了我们对 MOF 研究中合成可及性的理解,为该领域的未来进展提供了有价值的见解。
更新日期:2024-10-31
中文翻译:
MOFSynth:用于 MOF 合成似然预测的计算工具
在过去十年中,材料的高通量计算研究显着增加,这主要是由于计算机能力的进步,并引起了人们的极大兴趣。在金属有机框架 (MOF) 领域,已经用计算机设计了超过 100 万个假设的 MOF,但其中只有一小部分被合成。为了验证计算假设结果并加速该领域的进展,迫切需要区分更有可能合成用于实际应用的 MOF。本研究对 MOF 的可合成可能性进行了全面调查,利用一种新颖的计算方法,该方法基于 MOF 结构内的接头构象与其分离的自由气体状态之间的能量和几何形状之间的差异,因为这两者都已被证明是影响 MOF 合成的关键因素。我们的用户友好型工具简化了可合成性评估,只需极少的计算化学专业知识。通过从数据库(包括 QMOF、CoRE MOF 和 ToBaCCo)中解构超过 40,000 个 MOF,我们分析了定义 MOF 晶胞内接头菌株的关键参数。我们的结果表明,QMOF 和 CoRE MOF 包含更有前途的合成候选者,而 ToBaCCo 由于材料未优化,表现出相对较差的可合成性可能性。通过广泛的分析,我们确定了高度可合成 MOF 的最佳接头候选者。由高 Pearson 和 Spearman 系数证实的跨数据库能量分布的一致趋势表明,有可能省略优化计算,从而显著降低计算成本。 这项研究强调了接头变形和能量差异的重要性,并增强了我们对 MOF 研究中合成可及性的理解,为该领域的未来进展提供了有价值的见解。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号