当前位置:
X-MOL 学术
›
Adv. Theory Simul.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Exploring Electronic and Conformational Attributes of an Organic Donor-Bridge-Acceptor Molecular System
Advanced Theory and Simulations ( IF 2.9 ) Pub Date : 2024-10-30 , DOI: 10.1002/adts.202301212 Nikolas Echeverri, Jose Dario Perea, Salvador Leon
Advanced Theory and Simulations ( IF 2.9 ) Pub Date : 2024-10-30 , DOI: 10.1002/adts.202301212 Nikolas Echeverri, Jose Dario Perea, Salvador Leon
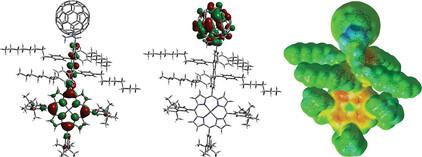
|
This research explores the electronic properties and conformational dynamics of the ZnP-COPV-
中文翻译:

探索有机供体-桥-受体分子系统的电子和构象属性
本研究通过专门的密度泛函理论 (DFT) 和分子动力学 (MD) 计算技术,探索了 ZnP-COPV- (锌卟啉 - 碳桥寡聚苯乙烯 - 富勒烯)有机半导体的电子特性和构象动力学。首先,通过 DFT 计算,仔细剖析基本的 HOMO(最高占据分子轨道)、LUMO(最低未占据分子轨道)和分子静电势 (MEP) 电子属性,从而深入了解组成供体-受体部分之间错综复杂的电荷转移过程。此外,采用 MD 模拟来揭示分子系统的多方面构象灵活性和稳定性。构象属性的均方根偏差 (RMSD)、端到端距离和扭转角定量分析支持碳桥寡苯乙烯 (COPV) 分子线的 骨架平面和刚性构象。与最初扩展的结构和热化后的约束扭转运动(ZnP-COPV 和 COPV- 的内 )相比,这种最小的端到端距离变化(在 Å 内 )展示了 COPV 随着时间的推移保持结构刚度的能力,与有效 共轭的概念一致。最后,通过时间依赖性 (TD)-DFT 的视角,探讨了随着分子结构随时间变化,HOMO-LUMO 能隙的动态演变、TD-DFT 激发能量和振荡器强度。 观察到的能隙变化强调了分子在面对结构修饰时的适应性,暗示了结构稳定性和增强的电子特性之间的有趣联系。这项研究全面了解了有机半导体中构象动力学和电子属性之间错综复杂的相互作用,为为尖端光电应用设计稳定、高性能的材料提供了至关重要的定量见解,并有助于促进对可持续能源转换的集体理解。
更新日期:2024-10-30
中文翻译:
探索有机供体-桥-受体分子系统的电子和构象属性
本研究通过专门的密度泛函理论 (DFT) 和分子动力学 (MD) 计算技术,探索了 ZnP-COPV- (锌卟啉 - 碳桥寡聚苯乙烯 - 富勒烯)有机半导体的电子特性和构象动力学。首先,通过 DFT 计算,仔细剖析基本的 HOMO(最高占据分子轨道)、LUMO(最低未占据分子轨道)和分子静电势 (MEP) 电子属性,从而深入了解组成供体-受体部分之间错综复杂的电荷转移过程。此外,采用 MD 模拟来揭示分子系统的多方面构象灵活性和稳定性。构象属性的均方根偏差 (RMSD)、端到端距离和扭转角定量分析支持碳桥寡苯乙烯 (COPV) 分子线的 骨架平面和刚性构象。与最初扩展的结构和热化后的约束扭转运动(ZnP-COPV 和 COPV- 的内 )相比,这种最小的端到端距离变化(在 Å 内 )展示了 COPV 随着时间的推移保持结构刚度的能力,与有效 共轭的概念一致。最后,通过时间依赖性 (TD)-DFT 的视角,探讨了随着分子结构随时间变化,HOMO-LUMO 能隙的动态演变、TD-DFT 激发能量和振荡器强度。 观察到的能隙变化强调了分子在面对结构修饰时的适应性,暗示了结构稳定性和增强的电子特性之间的有趣联系。这项研究全面了解了有机半导体中构象动力学和电子属性之间错综复杂的相互作用,为为尖端光电应用设计稳定、高性能的材料提供了至关重要的定量见解,并有助于促进对可持续能源转换的集体理解。
































 京公网安备 11010802027423号
京公网安备 11010802027423号