当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Comparison of Methodologies for Absolute Binding Free Energy Calculations of Ligands to Intrinsically Disordered Proteins
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-10-28 , DOI: 10.1021/acs.jctc.4c00942 Michail Papadourakis, Zoe Cournia, Antonia S. J. S. Mey, Julien Michel
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-10-28 , DOI: 10.1021/acs.jctc.4c00942 Michail Papadourakis, Zoe Cournia, Antonia S. J. S. Mey, Julien Michel
|
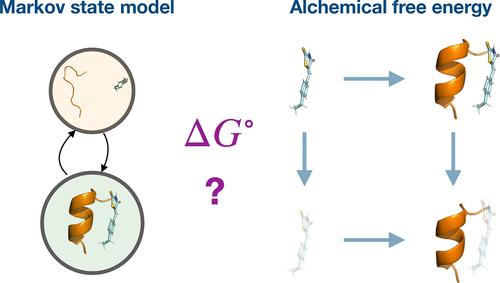
Modulating the function of Intrinsically Disordered Proteins (IDPs) with small molecules is of considerable importance given the crucial roles of IDPs in the pathophysiology of numerous diseases. Reported binding affinities for ligands to diverse IDPs vary broadly, and little is known about the detailed molecular mechanisms that underpin ligand efficacy. Molecular simulations of IDP ligand binding mechanisms can help us understand the mode of action of small molecule inhibitors of IDP function, but it is still unclear how binding energies can be modeled rigorously for such a flexible class of proteins. Here, we compare alchemical absolute binding free energy calculations (ABFE) and Markov-State Modeling (MSM) protocols to model the binding of the small molecule 10058-F4 to a disordered peptide extracted from a segment of the oncoprotein c-Myc. The ABFE results produce binding energy estimates that are sensitive to the choice of reference structure. In contrast, the MSM results produce more reproducible binding energy estimates consistent with weak mM binding affinities and transient intermolecular contacts reported in the literature.
中文翻译:

配体与内在无序蛋白质的绝对结合自由能计算方法的比较
鉴于 IDP 在许多疾病的病理生理学中的关键作用,用小分子调节内在无序蛋白 (IDP) 的功能具有相当重要的意义。据报道,配体与不同 IDP 的结合亲和力差异很大,并且对支撑配体功效的详细分子机制知之甚少。IDP 配体结合机制的分子模拟可以帮助我们了解 IDP 功能的小分子抑制剂的作用模式,但目前尚不清楚如何为这样一类灵活的蛋白质严格建模结合能。在这里,我们比较了炼金术绝对结合自由能计算 (ABFE) 和马尔可夫态建模 (MSM) 方案,以模拟小分子 10058-F4 与从癌蛋白 c-Myc 片段中提取的无序肽的结合。ABFE 结果产生对参考结构的选择敏感的结合能估计。相比之下,MSM 结果产生更可重复的结合能估计值,与文献中报道的弱 mM 结合亲和力和瞬时分子间接触一致。
更新日期:2024-10-29
中文翻译:
配体与内在无序蛋白质的绝对结合自由能计算方法的比较
鉴于 IDP 在许多疾病的病理生理学中的关键作用,用小分子调节内在无序蛋白 (IDP) 的功能具有相当重要的意义。据报道,配体与不同 IDP 的结合亲和力差异很大,并且对支撑配体功效的详细分子机制知之甚少。IDP 配体结合机制的分子模拟可以帮助我们了解 IDP 功能的小分子抑制剂的作用模式,但目前尚不清楚如何为这样一类灵活的蛋白质严格建模结合能。在这里,我们比较了炼金术绝对结合自由能计算 (ABFE) 和马尔可夫态建模 (MSM) 方案,以模拟小分子 10058-F4 与从癌蛋白 c-Myc 片段中提取的无序肽的结合。ABFE 结果产生对参考结构的选择敏感的结合能估计。相比之下,MSM 结果产生更可重复的结合能估计值,与文献中报道的弱 mM 结合亲和力和瞬时分子间接触一致。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号