当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Data and Molecular Fingerprint-Driven Machine Learning Approaches to Halogen Bonding
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2024-10-29 , DOI: 10.1021/acs.jcim.4c01427 Daniel P. Devore, Kevin L. Shuford
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2024-10-29 , DOI: 10.1021/acs.jcim.4c01427 Daniel P. Devore, Kevin L. Shuford
|
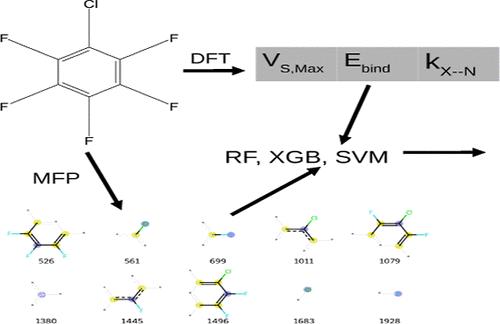
The ability to predict the strength of halogen bonds and properties of halogen bond (XB) donors has significant utility for medicinal chemistry and materials science. XBs are typically calculated through expensive ab initio methods. Thus, the development of tools and techniques for fast, accurate, and efficient property predictions has become increasingly more important. Herein, we employ three machine learning models to classify the XB donors and complexes by their principal halogen atom as well as predict the values of the maximum point on the electrostatic potential surface (VS,max) and interaction strength of the XB complexes through a molecular fingerprint and data-based analysis. The fingerprint analysis produces a root-mean-square error of ca. 7.5 and ca. 5.5 kcal mol–1 while predicting the VS,max for the halobenzene and haloethynylbenzene systems, respectively. However, the prediction of the binding energy between the XB donors and ammonia acceptor is shown to be within 1 kcal mol–1 of the density functional theory (DFT)-calculated energy. More accurate predictions can be made from the precalculated DFT data when compared to the fingerprint analysis.
中文翻译:

数据和分子指纹驱动的机器学习方法进行卤素键合
预测卤素键强度和卤素键 (XB) 供体性质的能力在药物化学和材料科学中具有重要用途。XB 通常是通过昂贵的 ab initio 方法计算的。因此,开发用于快速、准确和高效财产预测的工具和技术变得越来越重要。在此,我们采用三种机器学习模型,根据其主卤素原子对 XB 供体和配合物进行分类,并通过分子指纹和基于数据的分析预测静电势表面的最大值 (VS,max) 和 XB 配合物的相互作用强度。指纹图谱分析产生约 7.5 和约 5.5 kcal mol–1 的均方根误差,同时分别预测卤代苯和卤代乙炔苯系统的 VS,max。然而,对 XB 供体和氨受体之间结合能的预测显示在密度泛函理论 (DFT) 计算的能量的 1 kcal mol–1 以内。与指纹分析相比,预先计算的 DFT 数据可以做出更准确的预测。
更新日期:2024-10-29
中文翻译:
数据和分子指纹驱动的机器学习方法进行卤素键合
预测卤素键强度和卤素键 (XB) 供体性质的能力在药物化学和材料科学中具有重要用途。XB 通常是通过昂贵的 ab initio 方法计算的。因此,开发用于快速、准确和高效财产预测的工具和技术变得越来越重要。在此,我们采用三种机器学习模型,根据其主卤素原子对 XB 供体和配合物进行分类,并通过分子指纹和基于数据的分析预测静电势表面的最大值 (VS,max) 和 XB 配合物的相互作用强度。指纹图谱分析产生约 7.5 和约 5.5 kcal mol–1 的均方根误差,同时分别预测卤代苯和卤代乙炔苯系统的 VS,max。然而,对 XB 供体和氨受体之间结合能的预测显示在密度泛函理论 (DFT) 计算的能量的 1 kcal mol–1 以内。与指纹分析相比,预先计算的 DFT 数据可以做出更准确的预测。



















































 京公网安备 11010802027423号
京公网安备 11010802027423号