当前位置:
X-MOL 学术
›
Org. Chem. Front.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Mechanistic insight into the dehydrogenation reaction catalyzed by an MLC catalyst with dual ancillary ligand sites
Organic Chemistry Frontiers ( IF 4.6 ) Pub Date : 2024-10-29 , DOI: 10.1039/d4qo01767f Gui-Xiang Zhou, Cheng Hou
Organic Chemistry Frontiers ( IF 4.6 ) Pub Date : 2024-10-29 , DOI: 10.1039/d4qo01767f Gui-Xiang Zhou, Cheng Hou
|
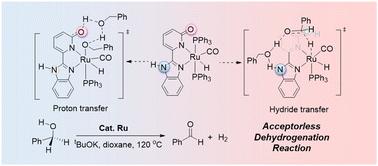
Metal–ligand cooperative (MLC) catalysis is central to modern catalytic chemistry, notable for its ability to enhance efficiency. Traditional MLC catalysts with a single auxiliary ligand site have limitations in optimizing catalytic activity, prompting increased interest in employing dual active sites for improved performance. In this study, density functional theory (DFT) is employed to explore the catalytic mechanism of a ruthenium complex featuring a 2-(2-benzimidazolyl)pyridine ligand in the dehydrogenation of benzyl alcohol. This catalyst is distinguished by its ability to use both the O–H group of hydroxy pyridine and the N–H group of benzimidazole as auxiliary sites during the reaction. The research uncovers a dynamic switching mechanism of ligand active sites across different catalytic stages. Specifically, the catalyst utilizes the oxygen site of the pyridone ligand as the proton acceptor during the proton transfer stage, while the N–H group of the benzimidazole ligand serves as the active site during the critical hydride transfer stage. This site-switching mechanism is elucidated through molecular plane parameter (MPP) analysis and Extended Transition State – Natural Orbitals for Chemical Valence (ETS-NOCV) analysis, which reveal that the N-site-assisted pathway during hydrogen transfer is characterized by reduced ligand deformation and enhanced orbital interactions. These factors contribute to the observed mechanistic selectivity. This dynamic site-switching strategy effectively lowers the reaction energy barrier and improves catalytic efficiency. The insights gained from this study not only clarify the roles of the ligand in various catalytic stages but also offer valuable theoretical guidance for the development of novel MLC catalysts with dual active sites.
中文翻译:

对具有双辅助配体位点的 MLC 催化剂催化的脱氢反应的机理见解
金属-配体合作 (MLC) 催化是现代催化化学的核心,以其提高效率的能力而著称。具有单个辅助配体位点的传统 MLC 催化剂在优化催化活性方面存在局限性,这促使人们越来越关注采用双活性位点来提高性能。在本研究中,密度泛函理论 (DFT) 用于探索具有 2-(2-苯并咪唑基)吡啶配体的钌配合物在苯甲醇脱氢过程中的催化机制。该催化剂的特点是能够在反应过程中同时使用羟基吡啶的 O-H 基团和苯并咪唑的 N-H 基团作为辅助位点。该研究揭示了不同催化阶段配体活性位点的动态切换机制。具体来说,催化剂在质子转移阶段利用吡啶酮配体的氧位点作为质子受体,而苯并咪唑配体的 N-H 基团在关键的氢化物转移阶段用作活性位点。这种位点切换机制通过分子平面参数 (MPP) 分析和扩展过渡态 - 化学价的自然轨道 (ETS-NOCV) 分析阐明,揭示了氢转移过程中 N 位点辅助途径的特点是配体变形减少和轨道相互作用增强。这些因素有助于观察到的机械选择性。这种动态位点切换策略有效地降低了反应能垒并提高了催化效率。本研究获得的见解不仅阐明了配体在各个催化阶段的作用,而且为开发具有双活性位点的新型 MLC 催化剂提供了有价值的理论指导。
更新日期:2024-11-01
中文翻译:
对具有双辅助配体位点的 MLC 催化剂催化的脱氢反应的机理见解
金属-配体合作 (MLC) 催化是现代催化化学的核心,以其提高效率的能力而著称。具有单个辅助配体位点的传统 MLC 催化剂在优化催化活性方面存在局限性,这促使人们越来越关注采用双活性位点来提高性能。在本研究中,密度泛函理论 (DFT) 用于探索具有 2-(2-苯并咪唑基)吡啶配体的钌配合物在苯甲醇脱氢过程中的催化机制。该催化剂的特点是能够在反应过程中同时使用羟基吡啶的 O-H 基团和苯并咪唑的 N-H 基团作为辅助位点。该研究揭示了不同催化阶段配体活性位点的动态切换机制。具体来说,催化剂在质子转移阶段利用吡啶酮配体的氧位点作为质子受体,而苯并咪唑配体的 N-H 基团在关键的氢化物转移阶段用作活性位点。这种位点切换机制通过分子平面参数 (MPP) 分析和扩展过渡态 - 化学价的自然轨道 (ETS-NOCV) 分析阐明,揭示了氢转移过程中 N 位点辅助途径的特点是配体变形减少和轨道相互作用增强。这些因素有助于观察到的机械选择性。这种动态位点切换策略有效地降低了反应能垒并提高了催化效率。本研究获得的见解不仅阐明了配体在各个催化阶段的作用,而且为开发具有双活性位点的新型 MLC 催化剂提供了有价值的理论指导。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号