当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Dependence of Thermally Activated Relaxation of Crystalline Stems on the Molecular Topology at Crystalline/Amorphous Interfaces in Polyethylene
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-10-28 , DOI: 10.1021/acs.jctc.4c00400 Yiyang Li, Jianlan Ye, Vipin Agrawal, Jay Oswald
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-10-28 , DOI: 10.1021/acs.jctc.4c00400 Yiyang Li, Jianlan Ye, Vipin Agrawal, Jay Oswald
|
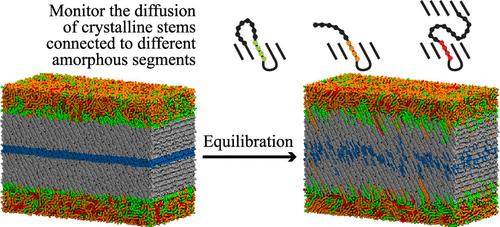
We investigate the relaxation dynamics of crystalline stems in relation to the molecular topology of the crystalline/amorphous interface, employing coarse–grained molecular dynamics. To efficiently generate model semicrystalline systems of linear polyethylene with a realistic interphase morphology, we simplified the Monte Carlo method by introducing molecular dynamics for faster relaxation. The structural properties of the generated systems are validated against experimental measurements, theoretical predictions, and existing simulation data. The models suggest that the probability distribution of loop-entry sites on the lamellar surface can be described by a power law in terms of the distance between the entry sites. By considering realistic interphase morphology, we are able to improve the prediction of the overall activation energy for the relaxation of crystalline stems, aligning it closely with experimental measurements. The largest model predicts that crystalline stems connected via large loops, i.e., those that exceed the entanglement length, and long tails are associated with increased activation energy; whereas stems connected to shorter tails show the lowest activation energy. These predictions can guide the future development of tougher semicrystalline polymers by providing insights into how amorphous chain morphology contributes to the activation energy and the relaxation dynamics of crystalline chains.
中文翻译:

结晶茎的热激活弛豫对聚乙烯中结晶/非晶界面分子拓扑结构的依赖性
我们采用粗晶分子动力学研究了结晶茎的弛豫动力学与结晶/非晶界面的分子拓扑结构的关系。为了有效地生成具有真实相间形态的线性聚乙烯模型半结晶系统,我们通过引入分子动力学来简化蒙特卡洛方法以加快弛豫速度。所生成系统的结构特性根据实验测量、理论预测和现有仿真数据进行验证。这些模型表明,层状表面上环入口位点的概率分布可以用幂律来描述,即入口位点之间的距离。通过考虑真实的界面形态,我们能够改进对结晶茎弛豫的总活化能的预测,使其与实验测量密切相关。最大的模型预测,通过大环连接的结晶茎,即那些超过缠结长度的结晶茎,以及长尾巴与增加的活化能有关;而连接到较短尾巴的茎显示出最低的活化能。这些预测可以通过深入了解非晶链形态如何影响结晶链的活化能和弛豫动力学来指导更坚韧的半结晶聚合物的未来发展。
更新日期:2024-10-28
中文翻译:
结晶茎的热激活弛豫对聚乙烯中结晶/非晶界面分子拓扑结构的依赖性
我们采用粗晶分子动力学研究了结晶茎的弛豫动力学与结晶/非晶界面的分子拓扑结构的关系。为了有效地生成具有真实相间形态的线性聚乙烯模型半结晶系统,我们通过引入分子动力学来简化蒙特卡洛方法以加快弛豫速度。所生成系统的结构特性根据实验测量、理论预测和现有仿真数据进行验证。这些模型表明,层状表面上环入口位点的概率分布可以用幂律来描述,即入口位点之间的距离。通过考虑真实的界面形态,我们能够改进对结晶茎弛豫的总活化能的预测,使其与实验测量密切相关。最大的模型预测,通过大环连接的结晶茎,即那些超过缠结长度的结晶茎,以及长尾巴与增加的活化能有关;而连接到较短尾巴的茎显示出最低的活化能。这些预测可以通过深入了解非晶链形态如何影响结晶链的活化能和弛豫动力学来指导更坚韧的半结晶聚合物的未来发展。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号