npj Computational Materials ( IF 9.4 ) Pub Date : 2024-10-24 , DOI: 10.1038/s41524-024-01424-1 Ebert Alvares, Kai Sellschopp, Bo Wang, ShinYoung Kang, Thomas Klassen, Brandon C. Wood, Tae Wook Heo, Paul Jerabek, Claudio Pistidda
|
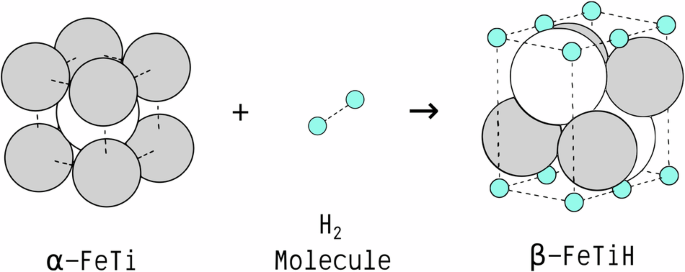
The quantification of interphase properties between metals and their corresponding hydrides is crucial for modeling the thermodynamics and kinetics of the hydrogenation processes in solid-state hydrogen storage materials. In particular, interphase boundary energies assume a pivotal role in determining the kinetics of nucleation, growth, and coarsening of hydrides, alongside accompanying morphological evolution during hydrogenation. The total interphase energy arises from both chemical bonding and mechanical strains in these solid-state systems. Since these contributions are usually coupled, it is challenging to distinguish via conventional computational approaches. Here, a comprehensive atomistic modeling methodology is developed to decouple chemical and mechanical energy contributions using first-principles calculations, of which feasibility is demonstrated by quantifying chemical and elastic strain energies of key interfaces within the FeTi metal-hydride system. Derived materials parameters are then employed for mesoscopic micromechanical analysis, predicting crystallographic orientations in line with experimental observations. The multiscale approach outlined verifies the importance of the chemo-mechanical interplay in the morphological evolution of growing hydride phases, and can be generalized to investigate other systems. In addition, it can streamline the design of atomistic models for the quantitative evaluation of interphase properties between dissimilar phases and allow for efficient predictions of their preferred phase boundary orientations.
中文翻译:
金属氢化物界面的多尺度建模——解耦化学机械能的量化
金属与其相应氢化物之间的相间特性的量化对于模拟固态储氢材料中加氢过程的热力学和动力学至关重要。特别是,相间边界能在确定氢化物的成核、生长和粗化动力学以及氢化过程中伴随的形态演变方面起着关键作用。总相间能来自这些固态系统中的化学键和机械应变。由于这些贡献通常是耦合的,因此通过传统的计算方法进行区分是具有挑战性的。在这里,开发了一种全面的原子建模方法,使用第一性原理计算来解耦化学能和机械能的贡献,通过量化 FeTi 金属氢化物系统中关键界面的化学能和弹性应变能来证明其可行性。然后,将衍生的材料参数用于细观微观力学分析,根据实验观察结果预测晶体取向。概述的多尺度方法验证了化学-机械相互作用在生长氢化物相的形态演变中的重要性,并且可以推广到研究其他系统。此外,它可以简化原子模型的设计,以定量评估不同相之间的相间特性,并允许有效预测它们的首选相边界方向。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号