Nature Metabolism ( IF 18.9 ) Pub Date : 2024-10-25 , DOI: 10.1038/s42255-024-01144-2 Shiyu Liu, Xiaojing Liu, Jason W. Locasale
|
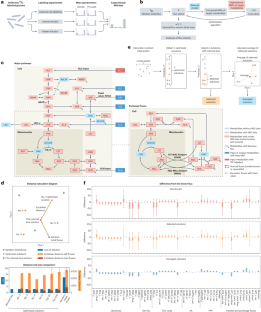
Metabolic flux analysis (MFA) is a computational approach to deciphering labelling patterns based on machine learning principles. Differing from typical machine learning algorithms that train a model from known datasets to make predictions, the commonly used MFA algorithm trains a metabolic network with data from isotope tracing experiments and directly outputs the learned information — that is, all fluxes in the network that best fit data3,5 (Fig. 1b). However, as a machine learning algorithm, current MFA methods often lack systematic evaluation and benchmarking, a standard practice in broader machine learning and artificial intelligence applications6. Issues such as algorithmic convergence, flux estimation accuracy and result robustness in MFA studies have been raised but remain largely unaddressed3, limiting the effectiveness and broader adoption of these automated tools in metabolic research.
To advance the capabilities of MFA for complex metabolic networks and extensive isotope tracing datasets, we developed an automated analysis methodology alongside a large-scale metabolic network model. This model comprises over 100 fluxes across key pathways, including glycolysis, the tricarboxylic acid (TCA) cycle, the pentose phosphate pathway (PPP), one-carbon metabolism, and several amino acid (AA) biosynthetic pathways (Fig. 1c, Supplementary Methods). Compared to contemporary MFA tools7,8,9, a notable feature of our methodology is the incorporation of organelle compartmentalization, facilitating accurate quantification of exchange fluxes between mitochondria and cytosol in eukaryotic cells (Fig. 1c). While other tools typically require tens of minutes to obtain a solution7,9, our methodology can generate an optimized solution, with fluxes that accurately explain the labelling pattern from a 13C tracing experiment on cultured cell lines, within 2 s on a desktop computer10 (Supplementary Fig. 1a–d). Nonetheless, a challenge arose from the observation that these optimized solutions could diverge significantly, showing considerable variability in certain net fluxes even with similar loss values (Fig. 1d, Supplementary Fig. 1e,f).To address this problem, we developed an optimization-averaging algorithm that refines the computation process by selecting a subset of solutions with minimal loss (selected solutions) from the pool of optimized solutions and averaging them to produce a new, more stable solution set (averaged solutions) (Fig. 1e, Supplementary Methods). These solutions, along with those generated using the typical strategy used in contemporary software (Supplementary Methods, Supplementary Fig. 1c), were benchmarked using simulated 13C tracing datasets generated from a known flux vector (Supplementary Fig. 2a). The results demonstrated that, relative to the benchmark, the optimization-averaging algorithm effectively reduced flux variability and improved the accuracy of the results in approximating the known flux, even with varying levels of data availability (Fig. 1f, Supplementary Figs. 2b–e and 3a–e).
中文翻译:
使用自动化方法从同位素示踪数据中定量代谢活性
代谢通量分析 (MFA) 是一种基于机器学习原理破译标记模式的计算方法。与从已知数据集训练模型进行预测的典型机器学习算法不同,常用的 MFA 算法使用来自同位素示踪实验的数据训练代谢网络,并直接输出学习到的信息,即网络中最适合数据的所有通量3,5(图 1b)。然而,作为一种机器学习算法,当前的 MFA 方法通常缺乏系统的评估和基准测试,而这是更广泛的机器学习和人工智能应用的标准做法6。MFA 研究中的问题包括算法收敛性、通量估计准确性和结果稳健性等问题,但在很大程度上仍未得到解决3,限制了这些自动化工具在代谢研究中的有效性和更广泛采用。
为了提高 MFA 对复杂代谢网络和广泛同位素追踪数据集的能力,我们开发了一种自动化分析方法以及大规模代谢网络模型。该模型包含跨关键途径的 100 多个通量,包括糖酵解、三羧酸 (TCA) 循环、磷酸戊糖途径 (PPP)、一碳代谢和几种氨基酸 (AA) 生物合成途径(图 1c,补充方法)。与当代 MFA 工具7,8,9 相比,我们方法的一个显着特点是结合了细胞器区室化,有助于准确量化真核细胞中线粒体和胞质溶胶之间的交换通量(图 1c)。虽然其他工具通常需要数十分钟才能获得溶液7,9,但我们的方法可以在 2 秒内在台式计算机上生成优化的解决方案,其通量可以准确解释培养细胞系上 13C 示踪实验的标记模式10(补充图 1a-d)。尽管如此,由于观察到这些优化的解决方案可能会显着发散,即使具有相似的损耗值,某些净磁通量也显示出相当大的变化(图 1d,补充图 1e,f)。为了解决这个问题,我们开发了一种优化平均算法,该算法通过从优化解决方案池中选择一个损失最小的解决方案子集(选定的解决方案)并对它们进行平均以产生一个新的、更稳定的解决方案集(平均解决方案)来改进计算过程(图 1e,补充方法)。这些解决方案,以及使用现代软件中使用的典型策略生成的解决方案(补充方法,补充图 1)。 1c),使用由已知磁通量矢量生成的模拟 13C 跟踪数据集进行基准测试(补充图 2a)。结果表明,相对于基准,优化-平均算法有效地降低了磁通量变化,并提高了结果在近似已知磁通量方面的准确性,即使数据可用性水平不同(图 1f,补充图 2b-e 和 3a-e)。






























 京公网安备 11010802027423号
京公网安备 11010802027423号