npj Computational Materials ( IF 9.4 ) Pub Date : 2024-10-24 , DOI: 10.1038/s41524-024-01433-0 Ryong-Gyu Lee, Yong-Hoon Kim
|
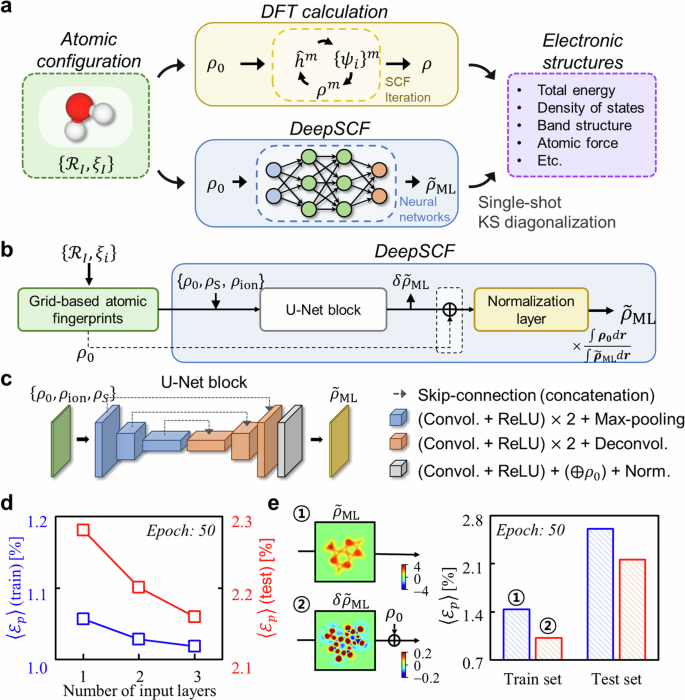
The self-consistent field (SCF) generation of the three-dimensional (3D) electron density distribution (ρ) represents a fundamental aspect of density functional theory (DFT) and related first-principles calculations, and how one can shorten or bypass the SCF loop represents a critical question in electronic structure theory from both practical and fundamental standpoints. Herein, a machine learning strategy, DeepSCF, is presented in which the map between the SCF ρ and the initial guess density (ρ0) constructed by the summation of neutral atomic densities is learned using 3D convolutional neural networks (CNNs). High accuracy and transferability of DeepSCF are achieved by first encoding ρ0 on a 3D grid and then expanding the input features to include atomic fingerprints beyond ρ0. The prediction of the residual density (δρ) rather than ρ itself is targeted, and given that δρ is indicative of chemical bonding information, a dataset of small-sized organic molecules featuring diverse bonding characters is adopted. The fidelity of DeepSCF is finally enhanced by subjecting the atomic geometries of the dataset to random rotations and strains. The effectiveness of DeepSCF is demonstrated using a complex carbon nanotube-based DNA sequencer model. This work evidences that the nearsightedness in electronic structure can be optimally represented via the spatial locality in CNNs, offering insight into the success of various machine learning-based atomistic materials simulations.
中文翻译:
通过网格投影原子指纹对自洽电子密度进行卷积网络学习
三维 (3D) 电子密度分布 (ρ) 的自洽场 (SCF) 生成代表了密度泛函论 (DFT) 和相关第一性原理计算的一个基本方面,如何从实践和基本角度缩短或绕过 SCF 回线代表了电子结构理论中的一个关键问题。在此,提出了一种机器学习策略 DeepSCF,其中 SCF ρ 与由中性原子密度总和构建的初始猜测密度 (ρ0) 之间的映射是使用 3D 卷积神经网络 (CNN) 学习的。DeepSCF 的高精度和可传递性是通过首先在 3D 网格上编码 ρ0,然后扩展输入特征以包括 ρ0 以外的原子指纹来实现的。预测残余密度 (δρ) 而不是 ρ 本身是有针对性的,鉴于 δρ 表示化学键合信息,因此采用了具有不同键合特性的小尺寸有机分子数据集。DeepSCF 的保真度最终通过使数据集的原子几何结构受到随机旋转和应变来增强。DeepSCF 的有效性使用基于碳纳米管的复杂 DNA 测序仪模型进行了验证。这项工作证明,电子结构的近视可以通过 CNN 中的空间局部性以最佳方式表示,从而为各种基于机器学习的原子材料模拟的成功提供见解。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号