Cell Death and Differentiation ( IF 13.7 ) Pub Date : 2024-10-24 , DOI: 10.1038/s41418-024-01390-7 Bo Kyoung Kim, Tatiana Goncharov, Sébastien A. Archaimbault, Filip Roudnicky, Joshua D. Webster, Peter D. Westenskow, Domagoj Vucic
|
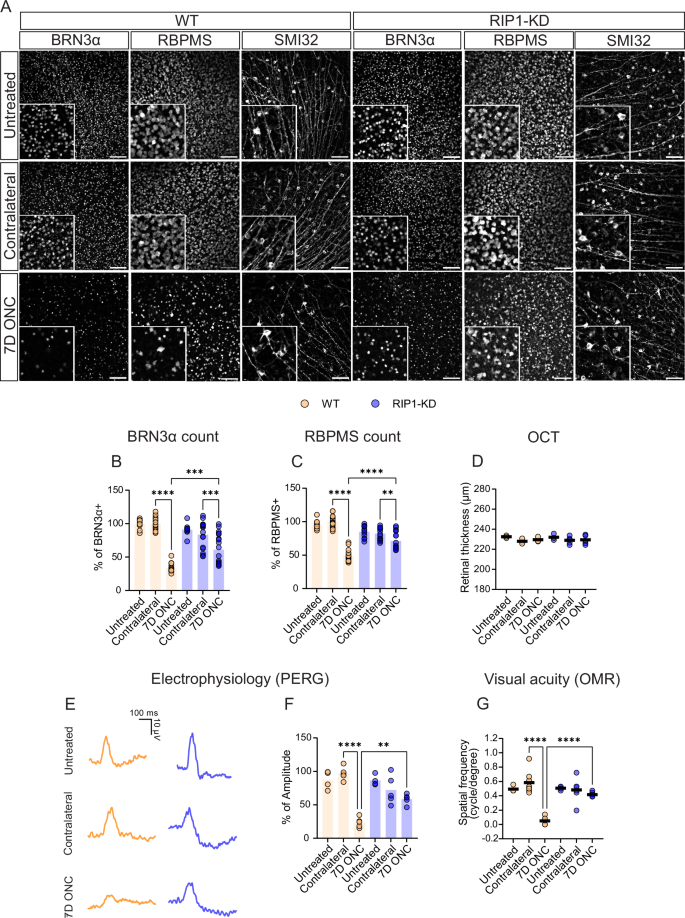
Receptor-interacting protein 1 (RIP1, RIPK1) is a critical mediator of multiple signaling pathways that promote inflammatory responses and cell death. The kinase activity of RIP1 contributes to the pathogenesis of a number of inflammatory and neurodegenerative diseases. However, the role of RIP1 in retinopathies remains unclear. This study demonstrates that RIP1 inhibition protects retinal ganglion cells (RGCs) in preclinical glaucoma models. Genetic inactivation of RIP1 improves RGC survival and preserves retinal function in the preclinical glaucoma models of optic nerve crush (ONC) and ischemia–reperfusion injury (IRI). In addition, the involvement of necroptosis in ONC and IRI glaucoma models was examined by utilizing RIP1 kinase-dead (RIP1-KD), RIP3 knockout (RIP3-KO), and MLKL knockout (MLKL-KO) mice. The number of RGCs, retinal thickness, and visual acuity were rescued in RIP1-kinase-dead (RIP1-KD) mice in both models, while wild-type (WT) mice experienced significant retinal thinning, RGC loss, and vision impairment. RIP3-KO and MLKL-KO mice showed moderate protective effects in the IRI model and limited in the ONC model. Furthermore, we confirmed that a glaucoma causative mutation in optineurin, OPTN-E50K, sensitizes cells to RIP1-mediated inflammatory cell death. RIP1 inhibition reduces RGC death and axonal degeneration following IRI in mice expressing OPTN-WT and OPTN-E50K variant mice. We demonstrate that RIP1 inactivation suppressed microglial infiltration in the RGC layer following glaucomatous damage. Finally, this study highlights that human glaucomatous retinas exhibit elevated levels of TNF and RIP3 mRNA and microglia infiltration, thus demonstrating the role of neuroinflammation in glaucoma pathogenesis. Altogether, these data indicate that RIP1 plays an important role in modulating neuroinflammation and that inhibiting RIP1 activity may provide a neuroprotective therapy for glaucoma.
中文翻译:
RIP1 抑制可保护眼损伤青光眼模型中的视网膜神经节细胞
受体相互作用蛋白 1 (RIP1, RIPK1) 是促进炎症反应和细胞死亡的多种信号通路的关键介质。RIP1 的激酶活性有助于许多炎症和神经退行性疾病的发病机制。然而,RIP1 在视网膜病变中的作用仍不清楚。本研究表明,RIP1 抑制可保护临床前青光眼模型中的视网膜神经节细胞 (RGC)。RIP1 的基因失活提高了 RGC 存活率并保留了视神经压碎 (ONC) 和缺血再灌注损伤 (IRI) 的临床前青光眼模型中的视网膜功能。此外,利用 RIP1 激酶死亡 (RIP1-KD) 、 RIP3 敲除 (RIP3-KO) 和 MLKL 敲除 (MLKL-KO) 小鼠检测坏死性凋亡对 ONC 和 IRI 青光眼模型的参与。在两种模型中,RIP1 激酶死亡 (RIP1-KD) 小鼠的 RGC 数量、视网膜厚度和视力均得到挽救,而野生型 (WT) 小鼠则出现明显的视网膜变薄、RGC 丢失和视力障碍。RIP3-KO 和 MLKL-KO 小鼠在 IRI 模型中表现出中等保护作用,而在 ONC 模型中表现出有限的保护作用。此外,我们证实 optineurin 中的青光眼致病突变 OPTN-E50K 使细胞对 RIP1 介导的炎症细胞死亡敏感。RIP1 抑制可减少表达 OPTN-WT 和 OPTN-E50K 变体小鼠的 IRI 后 RGC 死亡和轴突变性。我们证明 RIP1 失活抑制了青光眼损伤后 RGC 层中的小胶质细胞浸润。最后,这项研究强调,人类青光眼视网膜表现出 TNF 和 RIP3 mRNA 水平升高以及小胶质细胞浸润,从而证明了神经炎症在青光眼发病机制中的作用。 总而言之,这些数据表明 RIP1 在调节神经炎症中起重要作用,抑制 RIP1 活性可能为青光眼提供神经保护疗法。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号