当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Acceleration of Diffusion in Ab Initio Nanoreactor Molecular Dynamics and Application to Hydrogen Sulfide Oxidation
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-10-23 , DOI: 10.1021/acs.jctc.4c00826 Jan A. Meissner, Jan Meisner
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2024-10-23 , DOI: 10.1021/acs.jctc.4c00826 Jan A. Meissner, Jan Meisner
|
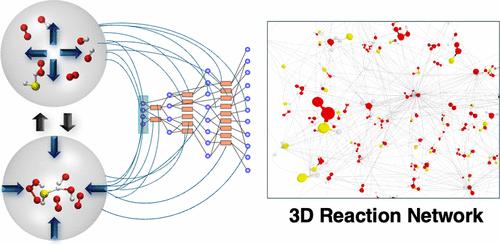
The computational description of chemical reactivity can become extremely complex when multiple different reaction products and intermediates come into play, forming a chemical reaction network. Therefore, computational methods for the automated construction of chemical reaction networks have been developed in the last decades. One of these methods, ab initio nanoreactor molecular dynamics (NMD), is based on external forces enhancing reactivity by e.g., periodically compressing the system and allowing it to relax. However, during the relaxation process, a significant simulation time is required to allow energy to dissipate and molecules to diffuse, making this part of the NMD simulation computationally intensive. This work aims to improve NMD by accelerating the diffusion process in the relaxation phase. We systematically investigate the speedup of reaction discovery gained by diffusion acceleration, leading to a factor of up to 28 in discovery frequency. Diffusion-accelerated nanoreactor molecular dynamics (DA-NMD) is then used to construct a reaction network of hydrogen sulfide oxidation under atmospheric conditions, where reactions are automatically detected by a change in the bond order and bond distance. A reaction network of 108 molecular species and 399 elementary reactions was constructed starting from hydrogen sulfide, hydroxy radicals, and molecular oxygen covering a broad variety of sulfur–oxygen chemistry and oxidation states of the sulfur atom ranging from −II to +VI.
中文翻译:

Ab Initio 纳米反应器中扩散加速分子动力学及其在硫化氢氧化中的应用
当多种不同的反应产物和中间体发挥作用,形成化学反应网络时,化学反应性的计算描述会变得极其复杂。因此,在过去的几十年中,已经开发了用于自动构建化学反应网络的计算方法。其中一种方法是 ab initio 纳米反应器分子动力学 (NMD),它基于外力,通过周期性压缩系统并使其松弛来增强反应性。然而,在弛豫过程中,需要大量的仿真时间才能让能量耗散和分子扩散,这使得 NMD 仿真的这一部分计算量很大。这项工作旨在通过加速弛豫阶段的扩散过程来改善 NMD。我们系统地研究了扩散加速获得的反应发现速度,导致发现频率高达 28 倍。然后使用扩散加速纳米反应器分子动力学 (DA-NMD) 在大气条件下构建硫化氢氧化反应网络,其中反应通过键序和键距离的变化自动检测。从硫化氢、羟基自由基和分子氧开始,构建了一个由 108 种分子和 399 种基本反应组成的反应网络,涵盖了硫原子的各种硫-氧化学和氧化态,范围从 −II 到 +VI。
更新日期:2024-10-24
中文翻译:
Ab Initio 纳米反应器中扩散加速分子动力学及其在硫化氢氧化中的应用
当多种不同的反应产物和中间体发挥作用,形成化学反应网络时,化学反应性的计算描述会变得极其复杂。因此,在过去的几十年中,已经开发了用于自动构建化学反应网络的计算方法。其中一种方法是 ab initio 纳米反应器分子动力学 (NMD),它基于外力,通过周期性压缩系统并使其松弛来增强反应性。然而,在弛豫过程中,需要大量的仿真时间才能让能量耗散和分子扩散,这使得 NMD 仿真的这一部分计算量很大。这项工作旨在通过加速弛豫阶段的扩散过程来改善 NMD。我们系统地研究了扩散加速获得的反应发现速度,导致发现频率高达 28 倍。然后使用扩散加速纳米反应器分子动力学 (DA-NMD) 在大气条件下构建硫化氢氧化反应网络,其中反应通过键序和键距离的变化自动检测。从硫化氢、羟基自由基和分子氧开始,构建了一个由 108 种分子和 399 种基本反应组成的反应网络,涵盖了硫原子的各种硫-氧化学和氧化态,范围从 −II 到 +VI。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号