当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Fragment and torsion biasing algorithms for construction of small organic molecules in proteins using DOCK
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2024-10-22 , DOI: 10.1002/jcc.27508 John D. Bickel, Brock T. Boysan, Robert C. Rizzo
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2024-10-22 , DOI: 10.1002/jcc.27508 John D. Bickel, Brock T. Boysan, Robert C. Rizzo
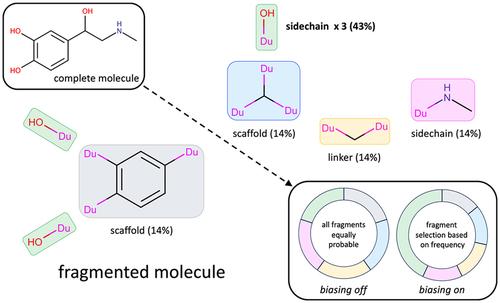
|
The computational construction of small organic molecules (de novo design), directly in a protein binding site, is an effective means for generating novel ligands tailored to fit the pocket environment. In this work, we present two new methods, which aim to improve de novo design outcomes using (1) biasing algorithms to prioritize selection and/or acceptance of fragments and torsions during growth, and (2) parallel-based clustering and pruning algorithms to remove duplicate molecules as candidate fragment are added. Large-scale testing encompassing thousands of simulations were employed to interrogate the methods in terms of multiple metrics which include numbers of duplicate molecules generated, pairwise-similarity, focused library reconstruction rates, fragment and torsion frequencies, fragment and torsion rank scores, interaction energy and drug-likeness scores, and 3D pose comparisons. The biasing algorithms, particularly those that include fragment and torsion components simultaneously, led to molecules that more closely mimicked the distributions of fragments and torsions found in drug-like libraries. The new parallel-based clustering and pruning algorithms, compared with the existing serial approach, also led to larger ensembles comprised of topologically unique molecules with much greater efficiency by removing redundant growth paths.
中文翻译:

使用 DOCK 构建蛋白质中有机小分子的片段和扭转偏倚算法
直接在蛋白质结合位点中计算构建有机小分子(从头设计)是生成适合口袋环境的新型配体的有效手段。在这项工作中,我们提出了两种新方法,旨在使用 (1) 偏置算法来改善从头设计结果,以优先考虑生长过程中片段和扭转的选择和/或接受,以及 (2) 基于并行的聚类和修剪算法以去除重复分子作为候选片段被添加。采用包含数千次模拟的大规模测试来根据多个指标来询问方法,包括生成的重复分子数量、成对相似性、聚焦库重建速率、片段和扭转频率、片段和扭转等级分数、相互作用能和药物相似性评分,以及 3D 姿势比较。偏倚算法,尤其是那些同时包含片段和扭转成分的算法,导致分子更接近于在药物样文库中发现的片段和扭转的分布。与现有的串行方法相比,新的基于并行的聚类和修剪算法还导致了由拓扑结构独特的分子组成的更大的集成,通过消除冗余的生长路径,效率更高。
更新日期:2024-10-22
中文翻译:
使用 DOCK 构建蛋白质中有机小分子的片段和扭转偏倚算法
直接在蛋白质结合位点中计算构建有机小分子(从头设计)是生成适合口袋环境的新型配体的有效手段。在这项工作中,我们提出了两种新方法,旨在使用 (1) 偏置算法来改善从头设计结果,以优先考虑生长过程中片段和扭转的选择和/或接受,以及 (2) 基于并行的聚类和修剪算法以去除重复分子作为候选片段被添加。采用包含数千次模拟的大规模测试来根据多个指标来询问方法,包括生成的重复分子数量、成对相似性、聚焦库重建速率、片段和扭转频率、片段和扭转等级分数、相互作用能和药物相似性评分,以及 3D 姿势比较。偏倚算法,尤其是那些同时包含片段和扭转成分的算法,导致分子更接近于在药物样文库中发现的片段和扭转的分布。与现有的串行方法相比,新的基于并行的聚类和修剪算法还导致了由拓扑结构独特的分子组成的更大的集成,通过消除冗余的生长路径,效率更高。






























 京公网安备 11010802027423号
京公网安备 11010802027423号