Advanced Synthesis & Catalysis ( IF 4.4 ) Pub Date : 2024-10-21 , DOI: 10.1002/adsc.202401170 Sara Gómez-Gil, Samuel Suárez-Pantiga, María R. Pedrosa, Roberto Sanz
|
Introduction
Nitroarenes are low-cost, stable, low-toxic, and abundant chemical raw materials, easily obtained through the straightforward electrophilic aromatic nitration of arenes. As one of the most accessible and economical nitrogen feedstock sources, the direct use of nitroaromatics to prepare value-added nitrogenated derivatives via various direct reductive C−N bond formation reactions, without prior reduction to the corresponding anilines,1 holds great significance and has seen a spectacular increase in interest in recent years.2 In contrast to using anilines for forming C−NAr bonds, the direct reductive coupling of nitroarenes demonstrates step economy and cost-effectiveness. Various reducing agents have been employed in such processes, including trivalent phosphorus reagents, organosilanes, carbon monoxide, boron compounds, hydrogen donors, reducing metals, metal carbonyls, visible light, or alcohols via hydrogen transfer reductions. Additionally, nitroarenes have also proven to be successful electrophiles for cross-coupling reactions.3
On the other hand, pyrroles are important N-heterocycles that form the core skeleton of various natural products and drugs with significant biological activities.4 They are also found in different forms of organic dyes and materials, including polymers and semiconductors.5 Although numerous methodologies have been developed for their synthesis and subsequent functionalization,6 the classical Paal-Knorr reaction remains one of the most prominent and widely applied method for synthesizing pyrroles from primary amines and 1,4-dicarbonyl compounds, making it a useful synthetic tool for accessing these heterocycles.7 Recent advances in this methodology have primarily focused on developing environmentally benign catalysts and conditions, as this reaction is known to be accelerated under protic or Lewis acidic environments.8 For the preparation of N-aryl pyrroles, anilines should be used as the nitrogenated counterpart (Scheme 1a). However, considering that anilines are typically obtained by the reduction of the corresponding readily available nitroarenes,1 the direct preparation of pyrroles from nitroarenes via cascade reactions in a single-pot operation is an attractive approach as it avoids the isolation of the corresponding anilines. The key issue for the success of this one-pot approach is the compatibility of the catalytic systems involved in the two steps of the cascade with both the reagents and the intermediates. Thus, the reducing step must be chemoselective for the nitro group, leaving the reducible dicarbonyl partner unaltered. Previously described processes in this area involve the use of large excesses of reducing metals, such as In or Sn, which generate substantial amounts of waste (Scheme 1b).9 Despite the synthetic potential of this approach for the preparation of pyrroles, this strategy has been demonstrated to be challenging and only very few metal-catalyzed procedures employing greener reducing agents have been reported. Most of them involve heterogeneous catalytic systems based on transition metals or catalysts that must be previously prepared by non-conventional procedures.10 Additionally, they require relatively harsh reaction conditions and the use of hydrogen or environmentally friendly formic acid as reductants (Scheme 1c). Very recently, Beller et al reported the first homogeneous non-noble metal catalyst for achieving this transformation,11a by applying a previously described protocol for the reduction of nitroarenes with formic acid as hydrogen donor and co-catalyst in the presence of an iron catalyst and a phosphorous-based ligand.11b Remarkably, this catalytic system shows good reactivity at room temperature and excellent chemoselectivity. However, although the Fe catalyst is commercially available, it requires the presence of Tetraphos as ligand and the use of a glove box for the manipulation of the reagents (Scheme 1d). In all the reported catalytic methods, hydrogen or hydrogen donors are employed, and the reactions take place via hydrogen atom transfer processes.

Synthesis of N-arylpyrroles from nitroarenes.
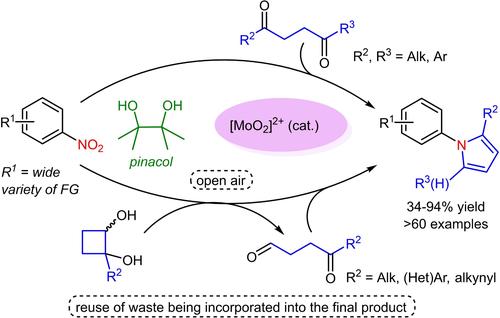
On the other hand, considering that molybdenum is the most abundant transition metal in seawater, serves as a cofactor for several enzymes, and is an essential trace element for human, animal and plant health, and is much less toxic than most heavy transition metals,12 we became interested in developing green synthetic methodologies involving redox processes using readily available dioxomolybdenum(VI) complexes.13 In this context, we previously described the selective reduction of nitroarenes with pinacol in the presence of other potentially reducible functional groups, such as carbonyls (Scheme 2a),14 and the oxidative cleavage of glycols with DMSO.15 In addition, we have combined these reactivities to prepare a wide variety of N-polyheterocycles, incorporating the waste reduction byproduct, a carbonyl derived from the initial reduction step, into the final compound (Scheme 2b).16 It is worth noting that the internal recycling of reaction waste for subsequent transformations is a highly relevant topic in organic synthesis. The use of a byproduct to facilitate or promote a downstream step was pioneered by Shibasaki,17 and since then, several groups have described sustainable processes based on the recycling of byproducts to promote or catalyze an array of subsequent transformations.18 However, fewer examples are known in which waste is recycled as reagent,19 and our group was the first time to describe the reuse of a waste byproduct as reactant for a downstream reaction, incorporating it into the final product.16 Since then, some other examples have been reported where a byproduct is embedded in the final product.20

Our previous work and this work.
In this context, we envisaged that the Mo-catalyzed chemoselective nitroarene reduction with pinacol, even in the presence of carbonyl groups, could be engaged with a subsequent cyclocondensation reaction with γ-dicarbonyls, enabling a cascade synthesis of pyrroles from nitroaromatics without involving hydrogenation processes. Alternatively, cyclobutane-1,2-diols could be used as reducing agents, with the subsequent incorporation of the waste reduction byproduct into the final heterocyclic moiety (Scheme 2c). Herein, we report our results exploiting this idea, which allows the direct synthesis of a variety of N-arylpyrroles from nitroaromatics.
中文翻译:
钼催化以乙二醇为还原剂从硝基芳烃直接合成吡咯
介绍
硝基芳烃是低成本、稳定、低毒、丰富的化工原料,很容易通过芳烃的直接亲电芳香族硝化获得。作为最容易获得和最经济的氮原料之一,直接使用硝基芳烃通过各种直接还原 C-N 键形成反应制备增值氮衍生物,而无需事先还原成相应的苯胺,1 具有重要意义,并且近年来引起了人们的极大兴趣。2 与使用苯胺形成 C-NAr 键相比,硝基芳烃的直接还原偶联表现出步骤经济性和成本效益。在此类工艺中采用了各种还原剂,包括三价磷试剂、有机硅烷、一氧化碳、硼化合物、氢供体、还原金属、金属羰基、可见光或通过氢转移还原的醇。此外,硝基芳烃也被证明是交叉偶联反应的成功亲电试剂。3
另一方面,吡咯是重要的 N-杂环,形成各种天然产物和具有重要生物活性的药物的核心骨架。4 它们也存在于不同形式的有机染料和材料中,包括聚合物和半导体。5 尽管已经开发了许多方法用于其合成和随后的功能化,6 但经典的 Paal-Knorr 反应仍然是从伯胺和 1,4-二羰基化合物合成吡咯的最突出和应用最广泛的方法之一,使其成为访问这些杂环的有用合成工具。7 该方法的最新进展主要集中在开发环境友好的催化剂和条件上,因为已知该反应在质子或 Lewis 酸性环境中会加速。8 对于N-芳基吡咯的制备,应使用苯胺作为氮化对应物(方案1a)。然而,考虑到苯胺通常是通过还原相应的现成硝基芳烃获得的,1 在单罐操作中通过级联反应从硝基芳烃中直接制备吡咯是一种有吸引力的方法,因为它避免了相应苯胺的分离。这种一锅法成功的关键问题是级联反应两个步骤中涉及的催化系统与试剂和中间体的兼容性。因此,还原步骤必须对硝基具有化学选择性,而可还原的二羰基伴侣保持不变。 前面描述的该领域的工艺涉及使用大量过量的还原金属,例如 In 或 Sn,这会产生大量废物(方案 1b)。9 尽管这种方法在制备吡咯方面具有合成潜力,但这种策略已被证明具有挑战性,并且只有极少数使用更环保还原剂的金属催化程序被报道。它们中的大多数涉及基于过渡金属或催化剂的非均相催化系统,这些系统必须事先通过非常规程序制备。10 此外,它们需要相对苛刻的反应条件,并使用氢气或环保的甲酸作为还原剂(方案 1c)。最近,Beller 等人报道了第一个实现这种转变的均相非贵金属催化剂,11a,方法是在铁催化剂和磷基配体存在下,应用先前描述的以甲酸作为氢供体和助催化剂还原亚硝基芳烃的方案。11b 值得注意的是,这种催化系统在室温下表现出良好的反应性和优异的化学选择性。然而,尽管 Fe 催化剂可以在市场上买到,但它需要存在 Tetraphos 作为配体,并使用手套箱来操作试剂(方案 1d)。在所有报道的催化方法中,都使用氢或氢供体,反应通过氢原子转移过程进行。
在图窗查看器PowerPoint 中打开
从硝基芳烃合成 N-芳基吡咯。
另一方面,考虑到钼是海水中含量最丰富的过渡金属,是多种酶的辅助因子,是人类、动物和植物健康所必需的微量元素,并且比大多数重过渡金属的毒性要小得多12,因此我们对使用现成的二氧代钼 (VI) 络合物开发涉及氧化还原工艺的绿色合成方法产生了兴趣。13 在此背景下,我们之前描述了在存在其他潜在可还原官能团(如羰基化合物)的情况下,用频哪醇选择性还原硝基芳烃(方案 2a)14 以及用 DMSO 对乙二醇进行氧化裂解。15 此外,我们还将这些反应性结合起来,制备了多种 N-多杂环,将减少废物的副产物(从初始还原步骤衍生的羰基化合物)掺入最终化合物中(方案 2b)。16 值得注意的是,反应废料的内部回收用于后续转化是有机合成中一个高度相关的主题。使用副产品来促进或促进下游步骤是由 Shibasaki 率先的,17 从那时起,几个小组描述了基于副产品回收的可持续过程,以促进或催化一系列后续转化。18 然而,已知的废物作为试剂回收的例子较少,19 我们小组首次描述了将废物副产物作为下游反应物的再利用,并将其纳入最终产品中。16 从那时起,还报道了其他一些副产品嵌入最终产品的例子。20
在图窗查看器PowerPoint 中打开
我们以前的工作和这次的工作。
在这种情况下,我们设想了即使在羰基存在的情况下,用 pinacol 进行 Mo 催化的化学选择性硝基芳烃还原也可以与随后的 γ-二羰基的环缩合反应进行,从而能够在不涉及氢化过程的情况下从硝基芳烃级联合成吡咯。或者,可以使用环丁烷-1,2-二醇作为还原剂,随后将减少废物的副产品掺入最终的杂环部分(方案 2c)。在此,我们报告了利用这一想法的结果,它允许从硝基芳烃直接合成各种 N-芳基吡咯。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号