当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Probing the performance of DFT in the structural characterization of [FeFe] hydrogenase models
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2024-10-17 , DOI: 10.1002/jcc.27515 Piotr Matczak, Philipp Buday, Stephan Kupfer, Helmar Görls, Grzegorz Mlostoń, Wolfgang Weigand
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2024-10-17 , DOI: 10.1002/jcc.27515 Piotr Matczak, Philipp Buday, Stephan Kupfer, Helmar Görls, Grzegorz Mlostoń, Wolfgang Weigand
|
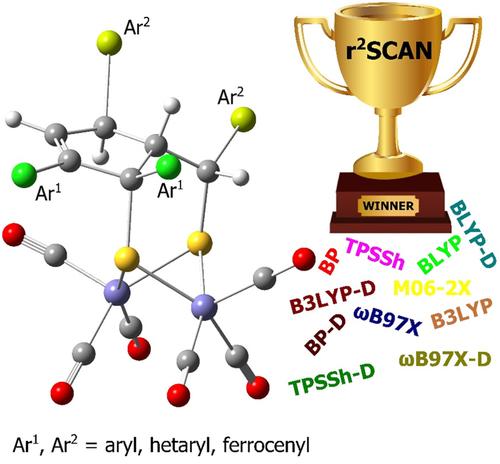
In this work, a series of DFT and DFT-D methods is combined with double-ζ basis sets to benchmark their performance in predicting the structures of five newly synthesized hexacarbonyl diiron complexes with a bridging ligand featuring a μ-S2C3 motif in a ring-containing unit functionalized with aromatic groups. Such complexes have been considered as [FeFe] hydrogenase catalytic site models with potential for eco-friendly energetic applications. According to this assessment, r2SCAN is identified as the density functional recommended for the reliable description of the molecular and crystal structures of the herein studied models. However, the butterfly (μ-S)2Fe2 core of the models demonstrates a minor deformation of its optimized geometry obtained from both molecular and periodic calculations. The FeFe bond length is slightly underestimated while the FeS bonds tend to be too long. Adding the D3(BJ) correction to r2SCAN does not lead to any improvement in the calculated structures.
中文翻译:

探索 DFT 在 [FeFe] 氢化酶模型结构表征中的性能
在这项工作中,将一系列 DFT 和 DFT-D 方法与双ζ基集相结合,以衡量它们在预测五种新合成的六羰基二铁配合物的结构方面的性能,这些配合物具有 μ-S 2C3 基序的桥接配体,在芳香族基团官能化的含环单元中。这种复合物被认为是 [FeFe] 氢化酶催化位点模型,具有环保能源应用的潜力。根据该评估,r2SCAN 被确定为推荐用于可靠描述本文研究模型的分子和晶体结构的密度泛函。然而,模型的蝶形 (μ-S)2Fe2 核心表现出从分子和周期计算中获得的优化几何形状的轻微变形。FeFe 键的长度略微低估,而 FeS 键往往太长。将 D3(BJ) 校正添加到 r2SCAN 不会导致计算结构的任何改进。
更新日期:2024-10-17
中文翻译:
探索 DFT 在 [FeFe] 氢化酶模型结构表征中的性能
在这项工作中,将一系列 DFT 和 DFT-D 方法与双ζ基集相结合,以衡量它们在预测五种新合成的六羰基二铁配合物的结构方面的性能,这些配合物具有 μ-S 2C3 基序的桥接配体,在芳香族基团官能化的含环单元中。这种复合物被认为是 [FeFe] 氢化酶催化位点模型,具有环保能源应用的潜力。根据该评估,r2SCAN 被确定为推荐用于可靠描述本文研究模型的分子和晶体结构的密度泛函。然而,模型的蝶形 (μ-S)2Fe2 核心表现出从分子和周期计算中获得的优化几何形状的轻微变形。FeFe 键的长度略微低估,而 FeS 键往往太长。将 D3(BJ) 校正添加到 r2SCAN 不会导致计算结构的任何改进。






























 京公网安备 11010802027423号
京公网安备 11010802027423号