Cell Research ( IF 28.1 ) Pub Date : 2024-10-17 , DOI: 10.1038/s41422-024-01027-x Ting Zhao, Xueying Guan, Yan Hu, Ziqian Zhang, Han Yang, Xiaowen Shi, Jin Han, Huan Mei, Luyao Wang, Lei Shao, Hongyu Wu, Qianqian Chen, Yongyan Zhao, Jiaying Pan, Yupeng Hao, Zeyu Dong, Xuan Long, Qian Deng, Shengjun Zhao, Mengke Zhang, Yumeng Zhu, Xiaowei Ma, Zequan Chen, Yayuan Deng, Zhanfeng Si, Xin Li, Tianzhen Zhang, Fei Gu, Xiaofeng Gu, Lei Fang
|
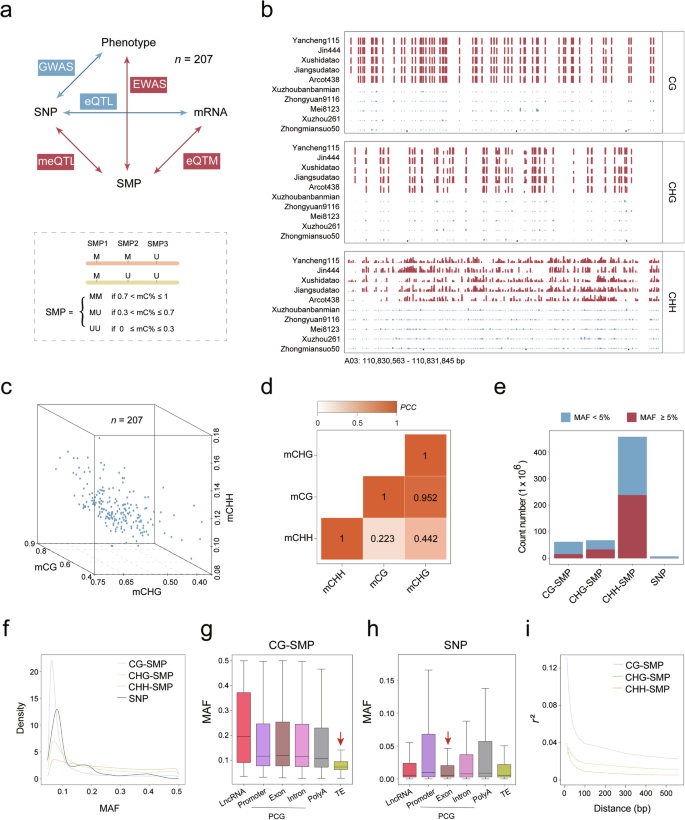
DNA methylation plays multiple regulatory roles in crop development. However, the relationships of methylation polymorphisms with genetic polymorphisms, gene expression, and phenotypic variation in natural crop populations remain largely unknown. Here, we surveyed high-quality methylomes, transcriptomes, and genomes obtained from the 20-days-post-anthesis (DPA) cotton fibers of 207 accessions and extended the classical framework of population genetics to epigenetics. Over 287 million single methylation polymorphisms (SMPs) were identified, 100 times more than the number of single nucleotide polymorphisms (SNPs). These SMPs were significantly enriched in intragenic regions while depleted in transposable elements. Association analysis further identified a total of 5,426,782 cis-methylation quantitative trait loci (cis-meQTLs), 5078 cis-expression quantitative trait methylation (cis-eQTMs), and 9157 expression quantitative trait loci (eQTLs). Notably, 36.39% of cis-eQTM genes were not associated with genetic variation, indicating that a large number of SMPs associated with gene expression variation are independent of SNPs. In addition, out of the 1715 epigenetic loci associated with yield and fiber quality traits, only 36 (2.10%) were shared with genome-wide association study (GWAS) loci. The construction of multi-omics regulatory networks revealed 43 cis-eQTM genes potentially involved in fiber development, which cannot be identified by GWAS alone. Among these genes, the role of one encoding CBL-interacting protein kinase 10 in fiber length regulation was successfully validated through gene editing. Taken together, our findings prove that DNA methylation data can serve as an additional resource for breeding purposes and can offer opportunities to enhance and expedite the crop improvement process.
中文翻译:
在 207 个棉花种质中,单核苷酸分辨率的全群 DNA 甲基化多态性揭示了对复杂性状的表观基因组贡献
DNA 甲基化在作物发育中起着多种调节作用。然而,甲基化多态性与自然作物种群中的遗传多态性、基因表达和表型变异的关系在很大程度上仍然未知。在这里,我们调查了从 207 个种质的开花后 20 天 (DPA) 棉纤维中获得的高质量甲基化组、转录组和基因组,并将群体遗传学的经典框架扩展到表观遗传学。鉴定出超过 2.87 亿个单甲基化多态性 (SMP),是单核苷酸多态性 (SNP) 数量的 100 倍。这些 SMP 在基因内区域显著富集,而在转座因子中耗尽。关联分析进一步确定了总共 5,426,782 个顺式甲基化数量性状位点 (cis-meQTL) 、5078 个顺式表达数量性状甲基化 (cis-eQTM) 和 9157 个表达数量性状位点 (eQTL)。值得注意的是,36.39% 的 cis-eQTM 基因与遗传变异无关,表明大量与基因表达变异相关的 SMPs 与 SNP 无关。此外,在与产量和纤维质量性状相关的 1715 个表观遗传位点中,只有 36 个 (2.10%) 与全基因组关联研究 (GWAS) 位点共享。多组学调控网络的构建揭示了 43 个 cis-eQTM 基因可能参与纤维发育,这些基因无法单独通过 GWAS 进行鉴定。在这些基因中,一个编码 CBL 相互作用蛋白激酶 10 的基因在纤维长度调节中的作用通过基因编辑成功验证。 综上所述,我们的研究结果证明 DNA 甲基化数据可以作为育种目的的额外资源,并且可以提供增强和加快作物改良过程的机会。



















































 京公网安备 11010802027423号
京公网安备 11010802027423号