Cell Death and Differentiation ( IF 13.7 ) Pub Date : 2024-10-09 , DOI: 10.1038/s41418-024-01398-z Anahita Ofoghi, Stefan Kotschi, Imke L. Lemmer, Daniel T. Haas, Nienke Willemsen, Batoul Bayer, Anna S. Jung, Sophie Möller, Stefanie Haberecht-Müller, Elke Krüger, Natalie Krahmer, Alexander Bartelt
|
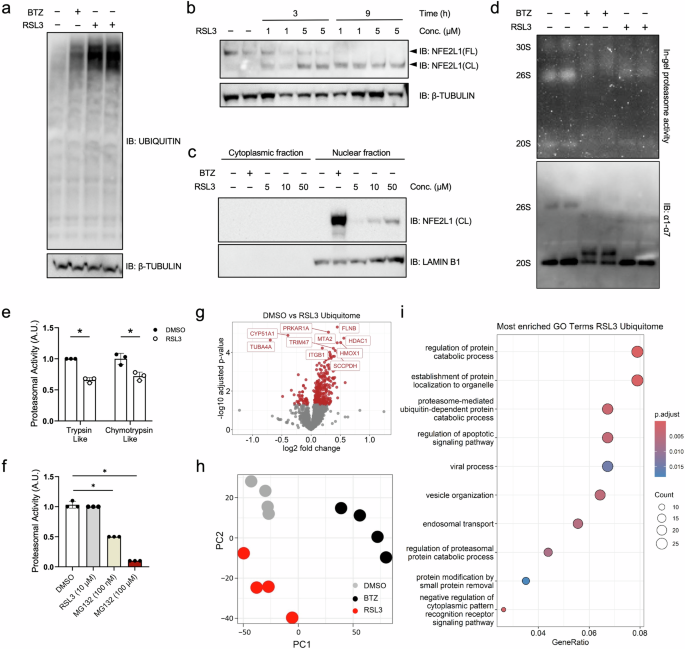
Ferroptosis is an iron-dependent, non-apoptotic form of cell death initiated by oxidative stress and lipid peroxidation. Recent evidence has linked ferroptosis to the action of the transcription factor Nuclear factor erythroid-2 derived,-like-1 (NFE2L1). NFE2L1 regulates proteasome abundance in an adaptive fashion, maintaining protein quality control to secure cellular homeostasis, but the regulation of NFE2L1 during ferroptosis and the role of the ubiquitin-proteasome system (UPS) herein are still unclear. In the present study, using an unbiased proteomic approach charting the specific ubiquitylation sites, we show that induction of ferroptosis leads to recalibration of the UPS. RSL3-induced ferroptosis inhibits proteasome activity and leads to global hyperubiquitylation, which is linked to NFE2L1 activation. As NFE2L1 resides in the endoplasmic reticulum tethered to the membrane, it undergoes complex posttranslational modification steps to become active and induce the expression of proteasome subunit genes. We show that proteolytic cleavage of NFE2L1 by the aspartyl protease DNA-damage inducible 1 homolog 2 (DDI2) is a critical step for the ferroptosis-induced feed-back loop of proteasome function. Cells lacking DDI2 cannot activate NFE2L1 in response to RSL3 and show global hyperubiquitylation. Genetic or chemical induction of ferroptosis in cells with a disrupted DDI2-NFE2L1 pathway diminishes proteasomal activity and promotes cell death. Also, treating cells with the clinical drug nelfinavir, which inhibits DDI2, sensitized cells to ferroptosis. In conclusion, our results provide new insight into the importance of the UPS in ferroptosis and highlight the role of the DDI2-NFE2L1 as a potential therapeutic target. Manipulating DDI2-NFE2L1 activity through chemical inhibition might help sensitizing cells to ferroptosis, thus enhancing existing cancer therapies.
中文翻译:
通过 DDI2 激活 NFE2L1-泛素-蛋白酶体系统可防止铁死亡
铁死亡是由氧化应激和脂质过氧化引发的铁依赖性、非凋亡性的细胞死亡形式。最近的证据表明,铁死亡与转录因子 erythroid-2 衍生的 -like-1 (NFE2L1) 的作用有关。NFE2L1 以适应性方式调节蛋白酶体丰度,维持蛋白质质量控制以确保细胞稳态,但铁死亡过程中 NFE2L1 的调节以及泛素-蛋白酶体系统 (UPS) 的作用仍不清楚。在本研究中,使用绘制特定泛素化位点的无偏蛋白质组学方法,我们表明铁死亡的诱导导致 UPS 的重新校准。RSL3 诱导的铁死亡抑制蛋白酶体活性并导致整体高泛素化,这与 NFE2L1 激活有关。由于 NFE2L1 位于拴在与膜相连的内质网中,因此它经历了复杂的翻译后修饰步骤,使其变得活跃并诱导蛋白酶体亚基基因的表达。我们表明,天冬氨酰蛋白酶 DNA 损伤诱导型 1 同源物 2 (DDI2) 对 NFE2L1 的蛋白水解切割是铁死亡诱导的蛋白酶体功能反馈环的关键步骤。缺乏 DDI2 的细胞不能响应 RSL3 激活 NFE2L1 并表现出整体高泛素化。在 DDI2-NFE2L1 通路中断的细胞中,铁死亡的遗传或化学诱导会降低蛋白酶体活性并促进细胞死亡。此外,用抑制 DDI2 的临床药物奈非那韦处理细胞,使细胞对铁死亡敏感。总之,我们的结果为 UPS 在铁死亡中的重要性提供了新的见解,并强调了 DDI2-NFE2L1 作为潜在治疗靶点的作用。 通过化学抑制操纵 DDI2-NFE2L1 活性可能有助于使细胞对铁死亡敏感,从而增强现有的癌症治疗。



















































 京公网安备 11010802027423号
京公网安备 11010802027423号