Nature Reviews Genetics ( IF 39.1 ) Pub Date : 2024-10-07 , DOI: 10.1038/s41576-024-00778-y Laura Harris, Ellen M. McDonagh, Xiaolei Zhang, Katherine Fawcett, Amy Foreman, Petr Daneck, Panagiotis I. Sergouniotis, Helen Parkinson, Francesco Mazzarotto, Michael Inouye, Edward J. Hollox, Ewan Birney, Tomas Fitzgerald
|
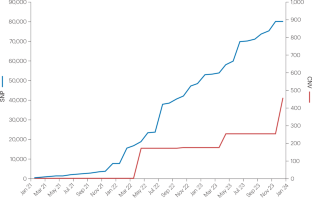
Decades of genetic association testing in human cohorts have provided important insights into the genetic architecture and biological underpinnings of complex traits and diseases. However, for certain traits, genome-wide association studies (GWAS) for common SNPs are approaching signal saturation, which underscores the need to explore other types of genetic variation to understand the genetic basis of traits and diseases. Copy number variation (CNV) is an important source of heritability that is well known to functionally affect human traits. Recent technological and computational advances enable the large-scale, genome-wide evaluation of CNVs, with implications for downstream applications such as polygenic risk scoring and drug target identification. Here, we review the current state of CNV-GWAS, discuss current limitations in resource infrastructure that need to be overcome to enable the wider uptake of CNV-GWAS results, highlight emerging opportunities and suggest guidelines and standards for future GWAS for genetic variation beyond SNPs at scale.
中文翻译:
超越 SNP 的全基因组关联检测
数十年来对人类队列的遗传关联测试为复杂性状和疾病的遗传结构和生物学基础提供了重要见解。然而,对于某些性状,常见 SNP 的全基因组关联研究 (GWAS) 正在接近信号饱和,这强调了探索其他类型的遗传变异以了解性状和疾病的遗传基础的必要性。拷贝数变异 (CNV) 是遗传性的重要来源,众所周知,它在功能上会影响人类特征。最近的技术和计算进步使 CNV 的大规模全基因组评估成为可能,对多基因风险评分和药物靶标识别等下游应用具有重要意义。在这里,我们回顾了 CNV-GWAS 的现状,讨论了当前需要克服的资源基础设施限制,以便更广泛地采用 CNV-GWAS 结果,突出了新出现的机会,并为未来 GWAS 的大规模 SNP 以外的遗传变异提出了指导方针和标准。





























 京公网安备 11010802027423号
京公网安备 11010802027423号