当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
An ANI‐2 enabled open‐source protocol to estimate ligand strain after docking
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2024-10-05 , DOI: 10.1002/jcc.27478 Francois Berenger, Koji Tsuda
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2024-10-05 , DOI: 10.1002/jcc.27478 Francois Berenger, Koji Tsuda
|
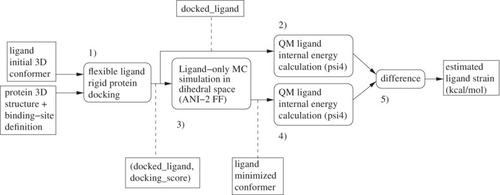
In protein‐ligand docking, the score assigned to a protein‐ligand complex is approximate. Especially, the internal energy of the ligand is difficult to compute precisely using a molecular mechanics based force‐field, introducing significant noise in the rank‐ordering of ligands. We propose an open‐source protocol (https://github.com/UnixJunkie/MMO ), using two quantum mechanics (QM) single point energy calculations, plus a Monte Carlo (Monte Carlo) based ligand minimization procedure in‐between, to estimate ligand strain after docking. The MC simulation uses the ANI‐2x (QM approximating) force field and is performed in the dihedral space. On some protein targets, using strain filtering after docking allows to significantly improve hit rates. We performed a structure‐based virtual screening campaign on nine protein targets from the Laboratoire d'Innovation Thérapeutique—PubChem assays dataset using Cambridge crystallographic data centre genetic optimization for ligand docking. Then, docked ligands were submitted to the strain estimation protocol and the impact on hit rate was analyzed. As for docking, the method does not always work. However, if sufficient active and inactive molecules are known for a given protein target, its efficiency can be evaluated.
中文翻译:

支持 ANI-2 的开源方案,用于估计对接后的配体菌株
在蛋白质-配体对接中,分配给蛋白质-配体复合物的分数是近似值。特别是,配体的内能很难使用基于分子力学的力场精确计算,从而在配体的等级排序中引入显着的噪声。我们提出了一种开源协议 (https://github.com/UnixJunkie/MMO),使用两个量子力学 (QM) 单点能量计算,加上一个基于蒙特卡洛 (Monte Carlo) 的配体最小化程序,以估计对接后的配体应变。MC 仿真使用 ANI‐2x(QM 近似)力场,并在二面体空间中执行。对于某些蛋白质靶标,在对接后使用菌株过滤可以显著提高命中率。我们使用剑桥晶体学数据中心对配体对接的遗传优化,对来自 Laboratoire d'Innovation Thérapeutique—PubChem 检测数据集的 9 个蛋白质靶标进行了基于结构的虚拟筛选活动。然后,将对接的配体提交给应变估计方案,并分析对命中率的影响。至于停靠,该方法并不总是有效。然而,如果已知给定蛋白质靶标有足够的活性和非活性分子,则可以评估其效率。
更新日期:2024-10-05
中文翻译:
支持 ANI-2 的开源方案,用于估计对接后的配体菌株
在蛋白质-配体对接中,分配给蛋白质-配体复合物的分数是近似值。特别是,配体的内能很难使用基于分子力学的力场精确计算,从而在配体的等级排序中引入显着的噪声。我们提出了一种开源协议 (https://github.com/UnixJunkie/MMO),使用两个量子力学 (QM) 单点能量计算,加上一个基于蒙特卡洛 (Monte Carlo) 的配体最小化程序,以估计对接后的配体应变。MC 仿真使用 ANI‐2x(QM 近似)力场,并在二面体空间中执行。对于某些蛋白质靶标,在对接后使用菌株过滤可以显著提高命中率。我们使用剑桥晶体学数据中心对配体对接的遗传优化,对来自 Laboratoire d'Innovation Thérapeutique—PubChem 检测数据集的 9 个蛋白质靶标进行了基于结构的虚拟筛选活动。然后,将对接的配体提交给应变估计方案,并分析对命中率的影响。至于停靠,该方法并不总是有效。然而,如果已知给定蛋白质靶标有足够的活性和非活性分子,则可以评估其效率。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号