当前位置:
X-MOL 学术
›
Phys. Rev. Lett.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Self-Consistent Determination of Single-Impurity Anderson Model Using Hybrid Quantum-Classical Approach on a Spin Quantum Simulator
Physical Review Letters ( IF 8.1 ) Pub Date : 2024-10-01 , DOI: 10.1103/physrevlett.133.140602 Xinfang Nie, Xuanran Zhu, Yu-ang Fan, Xinyue Long, Hongfeng Liu, Keyi Huang, Cheng Xi, Liangyu Che, Yuxuan Zheng, Yufang Feng, Xiaodong Yang, Dawei Lu
Physical Review Letters ( IF 8.1 ) Pub Date : 2024-10-01 , DOI: 10.1103/physrevlett.133.140602 Xinfang Nie, Xuanran Zhu, Yu-ang Fan, Xinyue Long, Hongfeng Liu, Keyi Huang, Cheng Xi, Liangyu Che, Yuxuan Zheng, Yufang Feng, Xiaodong Yang, Dawei Lu
|
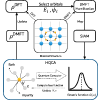
The accurate determination of the electronic structure of strongly correlated materials using first principle methods is of paramount importance in condensed matter physics, computational chemistry, and material science. However, due to the exponential scaling of computational resources, incorporating such materials into classical computation frameworks becomes prohibitively expensive. In 2016, Bauer et al. proposed a hybrid quantum-classical approach to correlated materials [B. Bauer et al., Hybrid quantum-classical approach to correlated materials, Phys. Rev. X 6, 031045 (2016).] that can efficiently tackle the electronic structure of complex correlated materials. Here, we experimentally demonstrate that approach to tackle the computational challenges associated with strongly correlated materials. By seamlessly integrating quantum computation into classical computers, we address the most computationally demanding aspect of the calculation, namely the computation of the Green’s function, using a spin quantum processor. Furthermore, we realize a self-consistent determination of the single impurity Anderson model through a feedback loop between quantum and classical computations. A quantum phase transition in the Hubbard model from the metallic phase to the Mott insulator is observed as the strength of electron correlation increases. As the number of qubits with high control fidelity continues to grow, our experimental findings pave the way for solving even more complex models, such as strongly correlated crystalline materials or intricate molecules.
中文翻译:

在自旋量子模拟器上使用混合量子经典方法自洽测定单杂质 Anderson 模型
使用第一性原理方法准确测定强相关材料的电子结构在凝聚态物理学、计算化学和材料科学中至关重要。然而,由于计算资源的指数级扩展,将这些材料整合到经典计算框架中变得非常昂贵。2016 年,Bauer 等 人提出了一种混合量子-经典相关材料的方法 [B. Bauer 等人,混合量子-经典方法,Phys. Rev. X6, 031045 (2016)],可以有效地处理复杂相关材料的电子结构。在这里,我们通过实验证明了这种方法可以解决与强相关材料相关的计算挑战。通过将量子计算无缝集成到经典计算机中,我们解决了计算中计算要求最高的方面,即使用自旋量子处理器计算格林函数。此外,我们通过量子计算和经典计算之间的反馈回路实现了对单个杂质 Anderson 模型的自洽确定。随着电子相关强度的增加,观察到哈伯德模型中从金属相到 Mott 绝缘体的量子相变。随着具有高控制保真度的量子比特数量不断增长,我们的实验结果为解决更复杂的模型铺平了道路,例如强相关的晶体材料或复杂的分子。
更新日期:2024-10-03
中文翻译:
在自旋量子模拟器上使用混合量子经典方法自洽测定单杂质 Anderson 模型
使用第一性原理方法准确测定强相关材料的电子结构在凝聚态物理学、计算化学和材料科学中至关重要。然而,由于计算资源的指数级扩展,将这些材料整合到经典计算框架中变得非常昂贵。2016 年,Bauer 等 人提出了一种混合量子-经典相关材料的方法 [B. Bauer 等人,混合量子-经典方法,Phys. Rev. X6, 031045 (2016)],可以有效地处理复杂相关材料的电子结构。在这里,我们通过实验证明了这种方法可以解决与强相关材料相关的计算挑战。通过将量子计算无缝集成到经典计算机中,我们解决了计算中计算要求最高的方面,即使用自旋量子处理器计算格林函数。此外,我们通过量子计算和经典计算之间的反馈回路实现了对单个杂质 Anderson 模型的自洽确定。随着电子相关强度的增加,观察到哈伯德模型中从金属相到 Mott 绝缘体的量子相变。随着具有高控制保真度的量子比特数量不断增长,我们的实验结果为解决更复杂的模型铺平了道路,例如强相关的晶体材料或复杂的分子。














































 京公网安备 11010802027423号
京公网安备 11010802027423号