Cell Death and Differentiation ( IF 13.7 ) Pub Date : 2024-10-03 , DOI: 10.1038/s41418-024-01385-4 Rui Ni, Ting Cao, Xiaoyun Ji, Angel Peng, Zhuxu Zhang, Guo-Chang Fan, Peter Stathopulos, Subrata Chakrabarti, Zhaoliang Su, Tianqing Peng
|
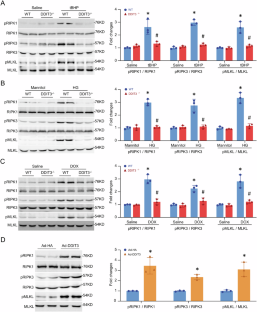
DNA damage-inducible transcript 3 (DDIT3) is a well-known transcription factor that regulates the expression of apoptosis-related genes for promoting apoptosis during endoplasmic reticulum stress. Here, we report an unrecognized role of DDIT3 in facilitating necroptosis. DDIT3 directly binds and competitively prevents the p38 MAPK-MK2 interaction and thereby blocking MK2 activation while stimulating p38 MAPK activation. This blockage of MK2 activation initially prevents RIPK1 phosphorylation at Ser320 (inactivation), subsequently relieving its suppression of RIPK1 activation. Consequently, p38 MAPK facilitates RIPK1 phosphorylation at Ser166 (activation) through DDIT3 phosphorylation-related mechanisms, leading to necroptosis. Mechanistically, a 10-amino acid segment (Glu19-Val28) within DDIT3’s N-terminus is identified to account for its pro-necroptotic function. In vivo studies demonstrate that forced expression of DDIT3 induces necroptosis, whereas deletion of DDIT3 alleviates necroptosis in mouse hearts under stress. These findings shed light on a novel regulatory mechanism by which DDIT3 promotes RIPK1 activation and subsequent necroptosis.
中文翻译:
DNA 损伤诱导转录本 3 正向调节 RIPK1 介导的坏死性凋亡
DNA 损伤诱导转录本 3 (DDIT3) 是一种众所周知的转录因子,可调节细胞凋亡相关基因的表达,从而在内质网应激期间促进细胞凋亡。在这里,我们报道了 DDIT3 在促进坏死性凋亡中未被认识的作用。DDIT3 直接结合并竞争性阻止 p38 MAPK-MK2 相互作用,从而阻断 MK2 激活,同时刺激 p38 MAPK 激活。这种 MK2 激活的阻断最初阻止了 RIPK1 在 Ser320 位点的磷酸化(失活),随后解除了其对 RIPK1 激活的抑制。因此,p38 MAPK 通过 DDIT3 磷酸化相关机制促进 RIPK1 在 Ser166 位点的磷酸化(激活),从而导致坏死性凋亡。从机制上讲,DDIT3 N 端内的 10 个氨基酸片段 (Glu19-Val28) 被确定为解释其促坏死性凋亡功能。体内研究表明,DDIT3 的强制表达会诱导坏死性凋亡,而 DDIT3 的缺失会减轻应激下小鼠心脏的坏死性凋亡。这些发现揭示了 DDIT3 促进 RIPK1 激活和随后坏死性凋亡的新调节机制。






























 京公网安备 11010802027423号
京公网安备 11010802027423号