Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Evaluating protocols for reproducible targeted metabolomics by NMR
Analyst ( IF 3.6 ) Pub Date : 2024-10-02 , DOI: 10.1039/d4an01015a Darcy Cochran, Panteleimon G. Takis, James L. Alexander, Benjamin H. Mullish, Nick Powell, Julian R. Marchesi, Robert Powers
Analyst ( IF 3.6 ) Pub Date : 2024-10-02 , DOI: 10.1039/d4an01015a Darcy Cochran, Panteleimon G. Takis, James L. Alexander, Benjamin H. Mullish, Nick Powell, Julian R. Marchesi, Robert Powers
|
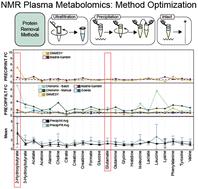
Metabolomics aims to study the downstream effects of variables like diet, environment, or disease on a given biological system. However, inconsistencies in sample preparation, data acquisition/processing protocols lead to reproducibility and accuracy concerns. A systematic study was conducted to assess how sample preparation methods and data analysis platforms affect metabolite susceptibility. A targeted panel of 25 metabolites was evaluated in 69 clinical metabolomics samples prepared following three different protocols: intact, ultrafiltration, and protein precipitation. The resulting metabolic profiles were characterized by 1D 1H nuclear magnetic resonance (NMR) spectroscopy and analyzed with Chenomx v8.3 and SMolESY software packages. Greater than 90% of the metabolites were extracted more efficiently using protein precipitation than filtration, which aligns with previously reported results. Additionally, analysis of data processing software suggests that metabolite concentrations were overestimated by Chenomx batch-fitting, which only appears reliable for determining relative fold changes rather than absolute quantification. However, an assisted-fit method provided sufficient guidance to achieve accurate results while avoiding a time-consuming fully manual-fitting approach. By combining our results with previous studies, we can now provide a list of 5 common metabolites [2-hydroxybutyrate (2-HB), choline, dimethylamine (DMA), glutamate, lactate] with a high degree of variability in reported fold changes and standard deviations that need careful consideration before being annotated as potential biomarkers. Our results show that sample preparation and data processing package critically impact clinical metabolomics study success. There is a clear need for an increased degree of standardization and harmonization of methods across the metabolomics community to ensure reliable outcomes.
中文翻译:

通过 NMR 评估可重复的靶向代谢组学方案
代谢组学旨在研究饮食、环境或疾病等变量对给定生物系统的下游影响。然而,样品制备、数据采集/处理方案的不一致会导致重现性和准确性问题。进行了一项系统研究,以评估样品制备方法和数据分析平台如何影响代谢物敏感性。在按照三种不同方案制备的 69 个临床代谢组学样品中评估了 25 种代谢物的靶向组:完整、超滤和蛋白质沉淀。通过 1D 1H 核磁共振 (NMR) 波谱表征所得代谢谱,并使用 Chenomx v8.3 和 SMolESY 软件包进行分析。与过滤相比,使用蛋白质沉淀法提取超过 90% 的代谢物的效率更高,这与之前报道的结果一致。此外,对数据处理软件的分析表明,Chenomx 批次拟合高估了代谢物浓度,这似乎仅可用于确定相对倍数变化而不是绝对定量。然而,辅助拟合方法提供了足够的指导来获得准确的结果,同时避免了耗时的完全手动拟合方法。通过将我们的结果与以前的研究相结合,我们现在可以提供 5 种常见代谢物 [2-羟基丁酸酯 (2-HB)、胆碱、二甲胺 (DMA)、谷氨酸、乳酸],这些代谢物在报告的倍数变化和标准差方面具有高度的可变性,在被注释为潜在生物标志物之前需要仔细考虑。我们的结果表明,样品制备和数据处理包对临床代谢组学研究的成功至关重要。 显然,整个代谢组学界需要提高方法的标准化和协调程度,以确保可靠的结果。
更新日期:2024-10-02
中文翻译:
通过 NMR 评估可重复的靶向代谢组学方案
代谢组学旨在研究饮食、环境或疾病等变量对给定生物系统的下游影响。然而,样品制备、数据采集/处理方案的不一致会导致重现性和准确性问题。进行了一项系统研究,以评估样品制备方法和数据分析平台如何影响代谢物敏感性。在按照三种不同方案制备的 69 个临床代谢组学样品中评估了 25 种代谢物的靶向组:完整、超滤和蛋白质沉淀。通过 1D 1H 核磁共振 (NMR) 波谱表征所得代谢谱,并使用 Chenomx v8.3 和 SMolESY 软件包进行分析。与过滤相比,使用蛋白质沉淀法提取超过 90% 的代谢物的效率更高,这与之前报道的结果一致。此外,对数据处理软件的分析表明,Chenomx 批次拟合高估了代谢物浓度,这似乎仅可用于确定相对倍数变化而不是绝对定量。然而,辅助拟合方法提供了足够的指导来获得准确的结果,同时避免了耗时的完全手动拟合方法。通过将我们的结果与以前的研究相结合,我们现在可以提供 5 种常见代谢物 [2-羟基丁酸酯 (2-HB)、胆碱、二甲胺 (DMA)、谷氨酸、乳酸],这些代谢物在报告的倍数变化和标准差方面具有高度的可变性,在被注释为潜在生物标志物之前需要仔细考虑。我们的结果表明,样品制备和数据处理包对临床代谢组学研究的成功至关重要。 显然,整个代谢组学界需要提高方法的标准化和协调程度,以确保可靠的结果。





























 京公网安备 11010802027423号
京公网安备 11010802027423号