当前位置:
X-MOL 学术
›
J. Am. Chem. Soc.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Reinforcement Learning for Improving Chemical Reaction Performance
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2024-10-02 , DOI: 10.1021/jacs.4c08866 Ajnabiul Hoque, Mihir Surve, Shivaram Kalyanakrishnan, Raghavan B. Sunoj
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2024-10-02 , DOI: 10.1021/jacs.4c08866 Ajnabiul Hoque, Mihir Surve, Shivaram Kalyanakrishnan, Raghavan B. Sunoj
|
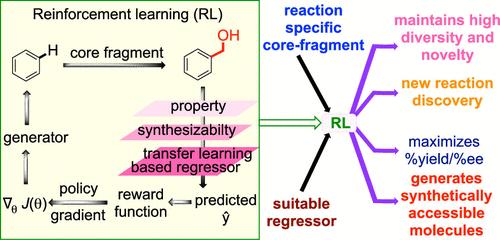
Deep learning (DL) methods have gained notable prominence in predictive and generative tasks in molecular space. However, their application in chemical reactions remains grossly underutilized. Chemical reactions are intrinsically complex: typically involving multiple molecules besides bond-breaking/forming events. In reaction discovery, one aims to maximize yield and/or selectivity that depends on a number of factors, mostly centered on reacting partners and reaction conditions. Herein, we introduce RE-EXPLORE, a novel approach that integrates deep reinforcement learning (RL) with an RNN-based deep generative model to identify prospective new reactants/catalysts, whose yield/selectivity is estimated using a pretrained regressor. Three chemical databases (ChEMBL, ZINC, and COCONUT containing half a million to one million unlabeled molecules) are independently used for pretraining the generators to enrich them with valuable information from diverse chemical space. Standard RL methods are found to be insufficient, as learners tend to prioritize exploitation for immediate gains, resulting in repetitive generation of same/similar molecules. Our engineered reward function includes a Tanimoto-based uniqueness factor within the RL loop that improved the exploration of the environment and has helped accrue larger returns. Integration of a user-defined core fragment into the generated molecules facilitated learning of specific reaction types. Together, RE-EXPLORE can navigate the reaction space toward practically meaningful regions and offers notable improvements across the three distinct reaction types considered in this study. It identifies high-yielding substrates and highly enantioselective chiral catalysts. This RL-based approach has the potential to expedite reaction discovery and aid in the synthesis planning of important compounds, including drugs and pharmaceuticals.
中文翻译:

用于提高化学反应性能的强化学习
深度学习 (DL) 方法在分子空间的预测和生成任务中获得了显着的重视。然而,它们在化学反应中的应用仍然严重未得到充分利用。化学反应本质上是复杂的:除了键断裂/形成事件外,通常还涉及多个分子。在反应发现中,其目标是最大限度地提高产量和/或选择性,这取决于许多因素,主要集中在反应伴侣和反应条件上。在此,我们介绍了 RE-EXPLORE,这是一种将深度强化学习 (RL) 与基于 RNN 的深度生成模型相结合的新方法,以识别潜在的新反应物/催化剂,其产率/选择性是使用预训练回归器估计的。三个化学数据库(ChEMBL、ZINC 和 COCONUT 包含五十万到一百万个未标记分子)独立用于预训练生成器,以使用来自不同化学空间的宝贵信息丰富它们。发现标准的 RL 方法是不够的,因为学习者倾向于优先考虑利用以获得即时收益,从而导致重复生成相同/相似的分子。我们设计的奖励函数在 RL 循环中包括一个基于 Tanimoto 的唯一性因子,该因子改善了对环境的探索并帮助积累了更大的回报。将用户定义的核心片段整合到生成的分子中,有助于了解特定的反应类型。总之,RE-EXPLORE 可以将反应空间引导到具有实际意义的区域,并在本研究中考虑的三种不同反应类型中提供显着改进。它鉴定高产率底物和高对映选择性手性催化剂。 这种基于 RL 的方法有可能加快反应发现并有助于重要化合物(包括药物和药物)的合成规划。
更新日期:2024-10-02
中文翻译:
用于提高化学反应性能的强化学习
深度学习 (DL) 方法在分子空间的预测和生成任务中获得了显着的重视。然而,它们在化学反应中的应用仍然严重未得到充分利用。化学反应本质上是复杂的:除了键断裂/形成事件外,通常还涉及多个分子。在反应发现中,其目标是最大限度地提高产量和/或选择性,这取决于许多因素,主要集中在反应伴侣和反应条件上。在此,我们介绍了 RE-EXPLORE,这是一种将深度强化学习 (RL) 与基于 RNN 的深度生成模型相结合的新方法,以识别潜在的新反应物/催化剂,其产率/选择性是使用预训练回归器估计的。三个化学数据库(ChEMBL、ZINC 和 COCONUT 包含五十万到一百万个未标记分子)独立用于预训练生成器,以使用来自不同化学空间的宝贵信息丰富它们。发现标准的 RL 方法是不够的,因为学习者倾向于优先考虑利用以获得即时收益,从而导致重复生成相同/相似的分子。我们设计的奖励函数在 RL 循环中包括一个基于 Tanimoto 的唯一性因子,该因子改善了对环境的探索并帮助积累了更大的回报。将用户定义的核心片段整合到生成的分子中,有助于了解特定的反应类型。总之,RE-EXPLORE 可以将反应空间引导到具有实际意义的区域,并在本研究中考虑的三种不同反应类型中提供显着改进。它鉴定高产率底物和高对映选择性手性催化剂。 这种基于 RL 的方法有可能加快反应发现并有助于重要化合物(包括药物和药物)的合成规划。





























 京公网安备 11010802027423号
京公网安备 11010802027423号