当前位置:
X-MOL 学术
›
Acc. Chem. Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Hydroxylamines: From Synthetic Intermediates to Synthetic Targets
Accounts of Chemical Research ( IF 16.4 ) Pub Date : 2024-09-30 , DOI: 10.1021/acs.accounts.4c00462 Roderick W. Bates
Accounts of Chemical Research ( IF 16.4 ) Pub Date : 2024-09-30 , DOI: 10.1021/acs.accounts.4c00462 Roderick W. Bates
|
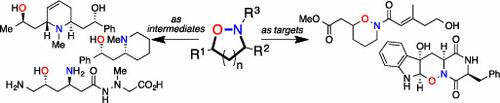
Synergy between the teaching and research activities of a University should be a source of new ideas, each informing the other. A classroom discussion gave rise to our concept of using hydroxylamines as a form of “tethered nitrogen” for alkaloid synthesis. The “tether” temporarily connects a nucleophilic nitrogen atom to the substrate, rendering an intermolecular reaction intramolecular, thus providing stereo- and regiochemical control for C–N bond formation. In the context of the synthesis of 1,3-amino alcohols, this necessitated the synthesis of isoxazolidines. This concept led to the exploration of methods for the synthesis of these heterocycles beyond the well-established 1,3-dipolar cycloadditions. The first two methods developed based upon this concept were palladium catalyzed cyclocarbonylation and silver catalyzed allene cyclization. These new methods were applied to two syntheses of sedamine and the synthesis of three Nuphar alkaloids. Intramolecular aza-Michael addition, combined with the use of cross-metathesis to generate the substrates, gave access to both isoxazolidines and tetrahydro-1,2-oxazines. This new method made possible a synthesis of monomorine with high stereochemical control. The extension to more complex alkaloids required the incorporation of additional chemistry. Combining allene cyclization with iminium ion chemistry allowed the extension to the more complex piperidine alkaloids porantheridine and sedinine. In these cases, successful stereocontrol relied on the ability to predict the conformation of the intermediates and the trajectory of the nucleophilic attack, which may be sterically or stereoelectronically controlled. Inspection of our collection of methods revealed that we had good methods for cis-3,5-disubstituted isoxazolidines and trans-3,6-disubstituted tetrahydro-1,2-oxazines. Inspired by earlier work involving iminium ions, methods were developed to provide the complementary trans-isoxazolidines and cis-oxazines, using an allylation reaction. This, combined with our interest in applications of hydroformylation, led to the synthesis of 5-hydroxysedamine. Venturing away from piperidines, this method was successfully applied to the synthesis of the β-amino acid antibiotic negamycin. Use of N,O-heterocycles as intermediates naturally sparked an interest in N,O-heterocycles as synthetic targets, as there are many natural products that contain the hydroxylamine moiety, often incorporated into an isoxazolidine or 1,2-oxazine ring. As the presence of the hydroxylamine moiety cannot be fully demonstrated using the usual spectroscopic methods, total synthesis can be employed to bridge this logical gap. A synthesis of raistrickindole A, utilizing an intramolecular Mitsunobu reaction of a hydroxamic acid to form a 1,2-oxazine, confirmed the structure of this diketopiperazine natural product. A synthesis of preisomide, using an aza-Michael addition to form the required 1,2-oxazine, also confirmed the structure of this natural product.
中文翻译:

羟胺:从合成中间体到合成靶标
大学教学和研究活动之间的协同作用应该是新思想的源泉,每个想法都为另一个想法提供信息。一次课堂讨论提出了我们使用羟胺作为生物碱合成的“栓系氮”的一种概念。“系绳”暂时将亲核氮原子连接到底物上,使分子间反应在分子内发生,从而为 C-N 键的形成提供立体和区域化学控制。在合成 1,3-氨基醇的背景下,这需要合成异恶唑烷。这一概念导致了对这些杂环合成方法的探索,超越了公认的 1,3-偶极环加成反应。基于这一概念开发的前两种方法是钯催化的环羰基化和银催化的烯丙烯环化。这些新方法应用于 sedamine 的两种合成和 3 种 Nuphar 生物碱的合成。分子内 aza-Michael 加成,结合使用交叉复分解生成底物,可以获得异恶唑烷和四氢-1,2-恶嗪。这种新方法使具有高立体化学控制的 monomorine 合成成为可能。扩展到更复杂的生物碱需要加入额外的化学成分。将烯丙烯环化与亚胺离子化学相结合,可以扩展到更复杂的哌啶生物碱 porantheridine 和 sedinine。在这些情况下,成功的立体控制依赖于预测中间体构象和亲核攻击轨迹的能力,这可能是空间或立体电子控制的。 检查我们收集的方法表明,我们有很好的方法来研究顺式-3,5-二取代的异噁唑烷和反式-3,6-二取代的四氢-1,2-恶嗪。受涉及亚胺离子的早期工作的启发,开发了使用烯丙基化反应提供互补反式异恶唑烷和顺式恶嗪的方法。这与我们对加氢甲酰化应用的兴趣相结合,导致了 5-羟基雪槲胺的合成。该方法远离哌啶类,成功地应用于 β-氨基酸抗生素 negamycin 的合成。使用 N,O-杂环作为中间体自然引发了人们对 N,O-杂环作为合成靶标的兴趣,因为有许多天然产物含有羟胺部分,通常掺入异恶唑胺或 1,2-恶嗪环中。由于使用通常的光谱方法无法完全证明羟胺部分的存在,因此可以采用全合成来弥合这一逻辑差距。利用异羟肟酸的分子内 Mitsunobu 反应形成 1,2-噁嗪的赖吲哚 A 的合成证实了这种二酮哌哌天然产物的结构。使用氮杂-迈克尔加成物形成所需的 1,2-恶嗪的 preisomide 合成也证实了这种天然产物的结构。
更新日期:2024-09-30
中文翻译:
羟胺:从合成中间体到合成靶标
大学教学和研究活动之间的协同作用应该是新思想的源泉,每个想法都为另一个想法提供信息。一次课堂讨论提出了我们使用羟胺作为生物碱合成的“栓系氮”的一种概念。“系绳”暂时将亲核氮原子连接到底物上,使分子间反应在分子内发生,从而为 C-N 键的形成提供立体和区域化学控制。在合成 1,3-氨基醇的背景下,这需要合成异恶唑烷。这一概念导致了对这些杂环合成方法的探索,超越了公认的 1,3-偶极环加成反应。基于这一概念开发的前两种方法是钯催化的环羰基化和银催化的烯丙烯环化。这些新方法应用于 sedamine 的两种合成和 3 种 Nuphar 生物碱的合成。分子内 aza-Michael 加成,结合使用交叉复分解生成底物,可以获得异恶唑烷和四氢-1,2-恶嗪。这种新方法使具有高立体化学控制的 monomorine 合成成为可能。扩展到更复杂的生物碱需要加入额外的化学成分。将烯丙烯环化与亚胺离子化学相结合,可以扩展到更复杂的哌啶生物碱 porantheridine 和 sedinine。在这些情况下,成功的立体控制依赖于预测中间体构象和亲核攻击轨迹的能力,这可能是空间或立体电子控制的。 检查我们收集的方法表明,我们有很好的方法来研究顺式-3,5-二取代的异噁唑烷和反式-3,6-二取代的四氢-1,2-恶嗪。受涉及亚胺离子的早期工作的启发,开发了使用烯丙基化反应提供互补反式异恶唑烷和顺式恶嗪的方法。这与我们对加氢甲酰化应用的兴趣相结合,导致了 5-羟基雪槲胺的合成。该方法远离哌啶类,成功地应用于 β-氨基酸抗生素 negamycin 的合成。使用 N,O-杂环作为中间体自然引发了人们对 N,O-杂环作为合成靶标的兴趣,因为有许多天然产物含有羟胺部分,通常掺入异恶唑胺或 1,2-恶嗪环中。由于使用通常的光谱方法无法完全证明羟胺部分的存在,因此可以采用全合成来弥合这一逻辑差距。利用异羟肟酸的分子内 Mitsunobu 反应形成 1,2-噁嗪的赖吲哚 A 的合成证实了这种二酮哌哌天然产物的结构。使用氮杂-迈克尔加成物形成所需的 1,2-恶嗪的 preisomide 合成也证实了这种天然产物的结构。



















































 京公网安备 11010802027423号
京公网安备 11010802027423号