Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Assessing AF2’s ability to predict structural ensembles of proteins
Structure ( IF 4.4 ) Pub Date : 2024-09-26 , DOI: 10.1016/j.str.2024.09.001 Jakob R. Riccabona, Fabian C. Spoendlin, Anna-Lena M. Fischer, Johannes R. Loeffler, Patrick K. Quoika, Timothy P. Jenkins, James A. Ferguson, Eva Smorodina, Andreas H. Laustsen, Victor Greiff, Stefano Forli, Andrew B. Ward, Charlotte M. Deane, Monica L. Fernández-Quintero
Structure ( IF 4.4 ) Pub Date : 2024-09-26 , DOI: 10.1016/j.str.2024.09.001 Jakob R. Riccabona, Fabian C. Spoendlin, Anna-Lena M. Fischer, Johannes R. Loeffler, Patrick K. Quoika, Timothy P. Jenkins, James A. Ferguson, Eva Smorodina, Andreas H. Laustsen, Victor Greiff, Stefano Forli, Andrew B. Ward, Charlotte M. Deane, Monica L. Fernández-Quintero
|
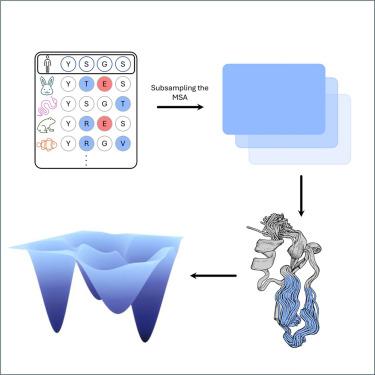
Recent breakthroughs in protein structure prediction have enhanced the precision and speed at which protein configurations can be determined. Additionally, molecular dynamics (MD) simulations serve as a crucial tool for capturing the conformational space of proteins, providing valuable insights into their structural fluctuations. However, the scope of MD simulations is often limited by the accessible timescales and the computational resources available, posing challenges to comprehensively exploring protein behaviors. Recently emerging approaches have focused on expanding the capability of AlphaFold2 (AF2) to predict conformational substates of protein. Here, we benchmark the performance of various workflows that have adapted AF2 for ensemble prediction and compare the obtained structures with ensembles obtained from MD simulations and NMR. We provide an overview of the levels of performance and accessible timescales that can currently be achieved with machine learning (ML) based ensemble generation. Significant minima of the free energy surfaces remain undetected.
中文翻译:

评估 AF2 预测蛋白质结构集合的能力
蛋白质结构预测的最新突破提高了确定蛋白质构型的精度和速度。此外,分子动力学 (MD) 模拟是捕获蛋白质构象空间的重要工具,为了解其结构波动提供了有价值的见解。然而,MD 模拟的范围通常受到可访问的时间尺度和可用计算资源的限制,这对全面探索蛋白质行为构成了挑战。最近新兴的方法侧重于扩展 AlphaFold2 (AF2) 预测蛋白质构象亚状态的能力。在这里,我们对将 AF2 用于集成预测的各种工作流程的性能进行了基准测试,并将获得的结构与从 MD 模拟和 NMR 获得的集成进行了比较。我们概述了当前可以通过基于机器学习 (ML) 的集成生成实现的性能级别和可访问的时间尺度。自由能表面的显着最小值仍未被检测到。
更新日期:2024-09-26
中文翻译:
评估 AF2 预测蛋白质结构集合的能力
蛋白质结构预测的最新突破提高了确定蛋白质构型的精度和速度。此外,分子动力学 (MD) 模拟是捕获蛋白质构象空间的重要工具,为了解其结构波动提供了有价值的见解。然而,MD 模拟的范围通常受到可访问的时间尺度和可用计算资源的限制,这对全面探索蛋白质行为构成了挑战。最近新兴的方法侧重于扩展 AlphaFold2 (AF2) 预测蛋白质构象亚状态的能力。在这里,我们对将 AF2 用于集成预测的各种工作流程的性能进行了基准测试,并将获得的结构与从 MD 模拟和 NMR 获得的集成进行了比较。我们概述了当前可以通过基于机器学习 (ML) 的集成生成实现的性能级别和可访问的时间尺度。自由能表面的显着最小值仍未被检测到。
































 京公网安备 11010802027423号
京公网安备 11010802027423号