当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Ultrafast dynamics in spatially confined photoisomerization: accelerated simulations through machine learning models
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2024-09-24 , DOI: 10.1039/d4cp01497a Weijia Xu, Haoyang Xu, Meifang Zhu, Jin Wen
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2024-09-24 , DOI: 10.1039/d4cp01497a Weijia Xu, Haoyang Xu, Meifang Zhu, Jin Wen
|
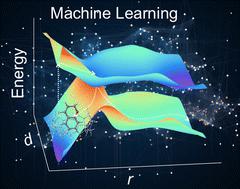
This study sheds light on the exploration of photoresponsive host–guest systems, highlighting the intricate interplay between confined spaces and photosensitive guest molecules. Conducting nonadiabatic molecular dynamics (NAMD) simulations based on electronic structure calculations for such large systems remains a formidable challenge. By leveraging machine learning (ML) as an accelerator for NAMD simulations, we analytically constructed excited-state potential energy surfaces along relevant collective variables to investigate photoisomerization processes efficiently. Combining the quantum mechanics/molecular mechanics (QM/MM) methodology with ML-based NAMD simulations, we elucidated the reaction pathways and identified the key degrees of freedom as reaction coordinates leading to conical intersections. A machine learning-based nonadiabatic dynamics model has been developed to compare the excited-state dynamics of the guest molecule, benzopyran, in both the gas phase and its behavior within the confined space of cucurbit[5]uril. This comparative analysis was designed to determine the influence of the environment on the photoisomerization rate of the guest molecule. The results underscore the effectiveness of ML models in simulating trajectory evolution in a cost-effective manner. This research offers a practical approach to accelerate NAMD simulations in large-scale systems of photochemical reactions, with potential applications in other host–guest complex systems.
中文翻译:

空间受限光异构化中的超快动力学:通过机器学习模型加速模拟
本研究阐明了光响应主客体系统的探索,突出了密闭空间和光敏客体分子之间错综复杂的相互作用。对此类大型系统进行基于电子结构计算的非绝热分子动力学 (NAMD) 仿真仍然是一项艰巨的挑战。通过利用机器学习 (ML) 作为 NAMD 模拟的加速器,我们沿着相关的集体变量分析构建了激发态势能表面,以有效地研究光异构化过程。将量子力学/分子力学 (QM/MM) 方法与基于 ML 的 NAMD 模拟相结合,我们阐明了反应途径,并将关键的自由度确定为导致圆锥交集的反应坐标。已经开发了一种基于机器学习的非绝热动力学模型,用于比较客体分子苯并吡咯在气相中的激发态动力学及其在葫芦[5]脲的密闭空间内的行为。该比较分析旨在确定环境对客体分子光异构化速率的影响。结果强调了 ML 模型以经济高效的方式模拟轨迹演变的有效性。这项研究提供了一种实用的方法,可以加速大规模光化学反应系统中的 NAMD 模拟,并可能应用于其他主客体复杂系统。
更新日期:2024-09-24
中文翻译:
空间受限光异构化中的超快动力学:通过机器学习模型加速模拟
本研究阐明了光响应主客体系统的探索,突出了密闭空间和光敏客体分子之间错综复杂的相互作用。对此类大型系统进行基于电子结构计算的非绝热分子动力学 (NAMD) 仿真仍然是一项艰巨的挑战。通过利用机器学习 (ML) 作为 NAMD 模拟的加速器,我们沿着相关的集体变量分析构建了激发态势能表面,以有效地研究光异构化过程。将量子力学/分子力学 (QM/MM) 方法与基于 ML 的 NAMD 模拟相结合,我们阐明了反应途径,并将关键的自由度确定为导致圆锥交集的反应坐标。已经开发了一种基于机器学习的非绝热动力学模型,用于比较客体分子苯并吡咯在气相中的激发态动力学及其在葫芦[5]脲的密闭空间内的行为。该比较分析旨在确定环境对客体分子光异构化速率的影响。结果强调了 ML 模型以经济高效的方式模拟轨迹演变的有效性。这项研究提供了一种实用的方法,可以加速大规模光化学反应系统中的 NAMD 模拟,并可能应用于其他主客体复杂系统。












































 京公网安备 11010802027423号
京公网安备 11010802027423号