当前位置:
X-MOL 学术
›
J. Phys. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Custom Hybrid Activation Function over Delocalization Error for Gap State Predictions of A2CeZrO6 (A = Ba2+, Sr2+, Ca2+, Mg2+) Proton Conductors: A First-Principles and Machine Learning Approach
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2024-09-24 , DOI: 10.1021/acs.jpcc.4c03379 D. Vignesh, Ela Rout
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2024-09-24 , DOI: 10.1021/acs.jpcc.4c03379 D. Vignesh, Ela Rout
|
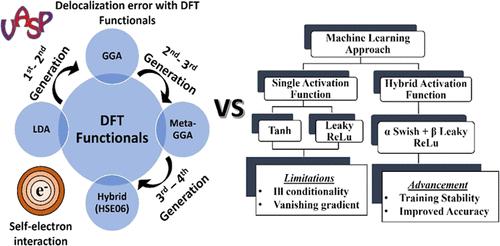
The crystal symmetry and band structure are two essential identifiers for a desirable proton-conducting electrolyte in fuel cell technology. However, structural and electrophysical inconsistency among cerium-based polymorphs originates due to multifacet issues ranging from cationic disorder (Ce4+ → Ce3+) to proton–polaron interaction. As a result, the distorted symmetry of Ca2CeZrO6 and Mg2CeZrO6 compounds displays an ambiguous trend in the forbidding gap due to differences in the spatial arrangement of atoms, chemical bonding, and hybridization. Besides material-driven limitations, the widely used density functional theory (DFT) simulations also face the band gap problem due to the lack of matrix element effects, final state effects, finite temperature offsets, and delocalization error by lower generation functionals (local density approximation (LDA) and generalized gradient approximation (GGA)). However, accurate estimations with higher rung functionals (hybrid (HSE06)) are often accompanied by critical limitations, including material-dependent response and exhaustive computations. Meanwhile, the sensitivity of an effective Hubbard parameter (Ueff) within the semiempirical DFT+U approach also limits their practical utility among complex oxides. As a result, we illustrate a mapping between the chemo-structural fingerprints and electronic band energy of distinct proton conductors using a state-of-the-art machine learning (ML) approach as a statistical learning and regression technique to combat the empirical approximations and derivative discontinuity of DFT simulations. In this study, we showcase the performance efficiency and limitations (training instability and generalization loss) of the designed artificial neural network (ANN) using a single activation function with a plausible solution via the custom hybrid activation function. According to our outcomes, the feature engineering of the hybrid activation function assists in overcoming numerical instability, ill conditionality, and dying gradient via adaptive neuro-controllers (tunable parameters) and nonlinear optimization of the loss landscape. As a result, the customized network was further used to model the potential energy surface (PES) of distinct proton conductors under their operating regimes (300–700 °C) to examine the proton landscape and transport characteristics with promising accuracy.
中文翻译:

A2CeZrO6 (A = Ba2+, Sr2+, Ca2+, Mg2+) 质子导体间隙态预测的去域误差上的自定义混合激活函数:第一性原理和机器学习方法
晶体对称性和能带结构是燃料电池技术中理想的质子导电电解质的两个基本标志。然而,基于铈的多晶型物之间的结构和电物理不一致是由于从阳离子无序 (Ce4+ → Ce3+) 到质子-极化子相互作用等多方面问题造成的。结果,由于原子空间布局、化学键合和杂化的差异,Ca2CeZrO6 和 Mg2CeZrO6 化合物的扭曲对称性在禁止间隙中表现出模糊趋势。除了材料驱动的限制外,广泛使用的密度泛函理论 (DFT) 仿真还面临着带隙问题,因为缺乏矩阵单元效应、最终状态效应、有限温度偏移以及低代泛函(局部密度近似 (LDA) 和广义梯度近似 (GGA))的离域误差。然而,具有更高梯级泛函的准确估计(混合 (HSE06))通常伴随着关键的局限性,包括材料依赖性响应和穷举计算。同时,半经验 DFT+U 方法中有效哈伯德参数 (Ueff) 的敏感性也限制了它们在复杂氧化物中的实际实用性。因此,我们使用最先进的机器学习 (ML) 方法作为统计学习和回归技术来对抗 DFT 模拟的经验近似和导数不连续性,说明了化学结构指纹和不同质子导体的电子带能量之间的映射。 在这项研究中,我们展示了所设计的人工神经网络 (ANN) 的性能效率和局限性(训练不稳定性和泛化损失),该网络使用单个激活函数,并通过自定义混合激活函数提供合理的解决方案。根据我们的结果,混合激活函数的特征工程通过自适应神经控制器(可调参数)和损失景观的非线性优化来帮助克服数值不稳定、病态条件和死亡梯度。因此,定制网络进一步用于模拟不同质子导体在其工作状态 (300-700 °C) 下的势能表面 (PES),以有希望的准确性检查质子景观和传输特性。
更新日期:2024-09-24
中文翻译:
A2CeZrO6 (A = Ba2+, Sr2+, Ca2+, Mg2+) 质子导体间隙态预测的去域误差上的自定义混合激活函数:第一性原理和机器学习方法
晶体对称性和能带结构是燃料电池技术中理想的质子导电电解质的两个基本标志。然而,基于铈的多晶型物之间的结构和电物理不一致是由于从阳离子无序 (Ce4+ → Ce3+) 到质子-极化子相互作用等多方面问题造成的。结果,由于原子空间布局、化学键合和杂化的差异,Ca2CeZrO6 和 Mg2CeZrO6 化合物的扭曲对称性在禁止间隙中表现出模糊趋势。除了材料驱动的限制外,广泛使用的密度泛函理论 (DFT) 仿真还面临着带隙问题,因为缺乏矩阵单元效应、最终状态效应、有限温度偏移以及低代泛函(局部密度近似 (LDA) 和广义梯度近似 (GGA))的离域误差。然而,具有更高梯级泛函的准确估计(混合 (HSE06))通常伴随着关键的局限性,包括材料依赖性响应和穷举计算。同时,半经验 DFT+U 方法中有效哈伯德参数 (Ueff) 的敏感性也限制了它们在复杂氧化物中的实际实用性。因此,我们使用最先进的机器学习 (ML) 方法作为统计学习和回归技术来对抗 DFT 模拟的经验近似和导数不连续性,说明了化学结构指纹和不同质子导体的电子带能量之间的映射。 在这项研究中,我们展示了所设计的人工神经网络 (ANN) 的性能效率和局限性(训练不稳定性和泛化损失),该网络使用单个激活函数,并通过自定义混合激活函数提供合理的解决方案。根据我们的结果,混合激活函数的特征工程通过自适应神经控制器(可调参数)和损失景观的非线性优化来帮助克服数值不稳定、病态条件和死亡梯度。因此,定制网络进一步用于模拟不同质子导体在其工作状态 (300-700 °C) 下的势能表面 (PES),以有希望的准确性检查质子景观和传输特性。












































 京公网安备 11010802027423号
京公网安备 11010802027423号