Nature Metabolism ( IF 18.9 ) Pub Date : 2024-09-23 , DOI: 10.1038/s42255-024-01139-z Thomas G. Hill, Rui Gao, Anna Benrick, Lakshmi Kothegala, Nils Rorsman, Cristiano Santos, Samuel Acreman, Linford J. Briant, Haiqiang Dou, Nikhil R. Gandasi, Claudia Guida, Elizabeth Haythorne, Marsha Wallace, Jakob G. Knudsen, Caroline Miranda, Johan Tolö, Anne Clark, Lucy Davison, Joachim Størling, Andrei Tarasov, Frances M. Ashcroft, Patrik Rorsman, Quan Zhang
|
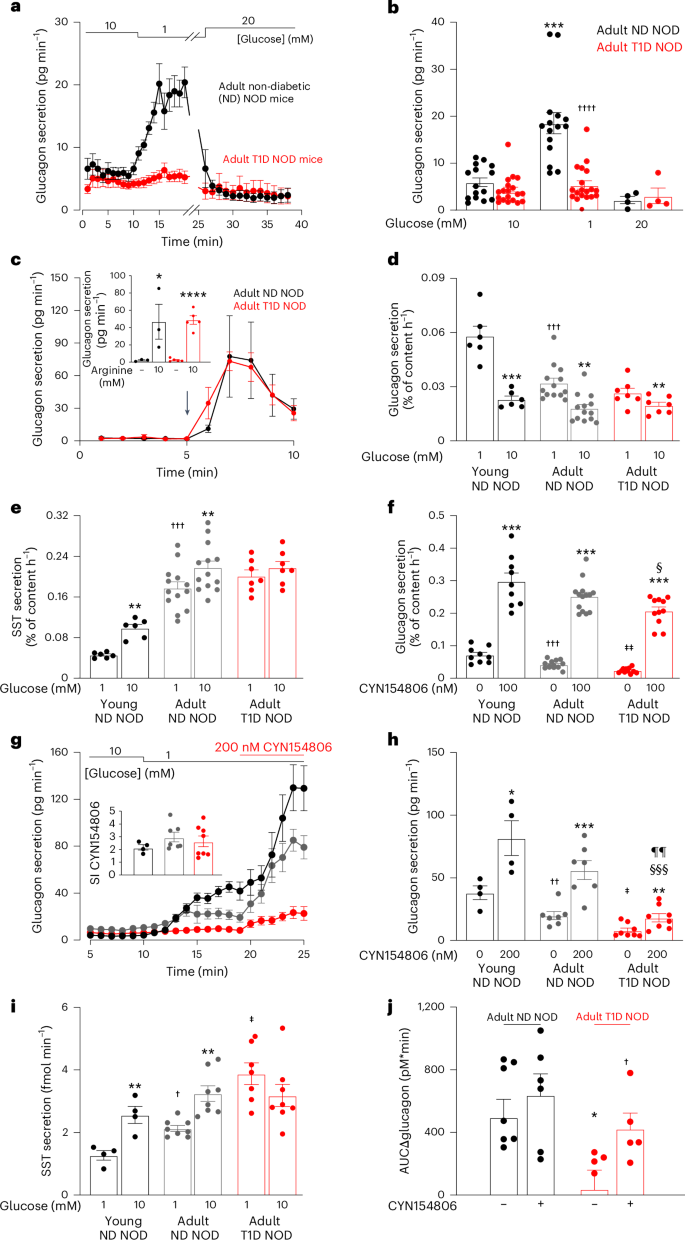
Diabetes mellitus involves both insufficient insulin secretion and dysregulation of glucagon secretion1. In healthy people, a fall in plasma glucose stimulates glucagon release and thereby increases counter-regulatory hepatic glucose production. This response is absent in many patients with type-1 diabetes (T1D)2, which predisposes to severe hypoglycaemia that may be fatal and accounts for up to 10% of the mortality in patients with T1D3. In rats with chemically induced or autoimmune diabetes, counter-regulatory glucagon secretion can be restored by SSTR antagonists4,5,6,7 but both the underlying cellular mechanism and whether it can be extended to humans remain unestablished. Here, we show that glucagon secretion is not stimulated by low glucose in isolated human islets from donors with T1D, a defect recapitulated in non-obese diabetic mice with T1D. This occurs because of hypersecretion of somatostatin, leading to aberrant paracrine inhibition of glucagon secretion. Normally, KATP channel-dependent hyperpolarization of β-cells at low glucose extends into the δ-cells through gap junctions, culminating in suppression of action potential firing and inhibition of somatostatin secretion. This ‘electric brake’ is lost following autoimmune destruction of the β-cells, resulting in impaired counter-regulation. This scenario accounts for the clinical observation that residual β-cell function correlates with reduced hypoglycaemia risk8.
中文翻译:
β 细胞与 δ 细胞耦合的丢失是 1 型糖尿病中低血糖诱导的胰高血糖素分泌受损的基础
糖尿病涉及胰岛素分泌不足和胰高血糖素分泌失调1。在健康人中,血浆葡萄糖的下降会刺激胰高血糖素的释放,从而增加反调节肝葡萄糖的产生。许多 1 型糖尿病 (T1D)2 患者不存在这种反应,易患严重低血糖,这可能是致命的,占 T1D3 患者死亡率的 10%。在患有化学诱导或自身免疫性糖尿病的大鼠中,SSTR 拮抗剂可以恢复反调节胰高血糖素的分泌4,5,6,7,但潜在的细胞机制以及它是否可以扩展到人类仍未确定。在这里,我们表明胰高血糖素分泌不受 T1D 供体离体人胰岛中低葡萄糖的刺激,这种缺陷在患有 T1D 的非肥胖糖尿病小鼠中概括了。这是因为生长抑素分泌过多,导致胰高血糖素分泌旁分泌异常抑制。正常情况下,低葡萄糖时 β 细胞的 KATP 通道依赖性超极化通过间隙连接延伸到 δ 细胞中,最终抑制动作电位放电和抑制生长抑素分泌。这种“电制动器”在 β 细胞自身免疫性破坏后丢失,导致反调节受损。这种情况解释了临床观察结果,即残余的 β 细胞功能与降低的低血糖风险相关8。



















































 京公网安备 11010802027423号
京公网安备 11010802027423号