Nature Machine Intelligence ( IF 18.8 ) Pub Date : 2024-09-18 , DOI: 10.1038/s42256-024-00900-z
Yuyan Ni , Shikun Feng , Xin Hong , Yuancheng Sun , Wei-Ying Ma , Zhi-Ming Ma , Qiwei Ye , Yanyan Lan
|
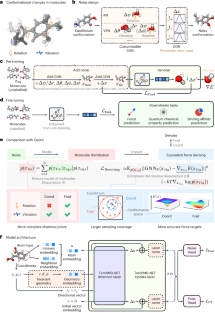
Deep learning methods have been considered promising for accelerating molecular screening in drug discovery and material design. Due to the limited availability of labelled data, various self-supervised molecular pre-training methods have been presented. Although many existing methods utilize common pre-training tasks in computer vision and natural language processing, they often overlook the fundamental physical principles governing molecules. In contrast, applying denoising in pre-training can be interpreted as an equivalent force learning, but the limited noise distribution introduces bias into the molecular distribution. To address this issue, we introduce a molecular pre-training framework called fractional denoising, which decouples noise design from the constraints imposed by force learning equivalence. In this way, the noise becomes customizable, allowing for incorporating chemical priors to substantially improve the molecular distribution modelling. Experiments demonstrate that our framework consistently outperforms existing methods, establishing state-of-the-art results across force prediction, quantum chemical properties and binding affinity tasks. The refined noise design enhances force accuracy and sampling coverage, which contribute to the creation of physically consistent molecular representations, ultimately leading to superior predictive performance.
中文翻译:
通过分数降噪进行预训练以增强分子特性预测
深度学习方法被认为有望加速药物发现和材料设计中的分子筛选。由于标记数据的可用性有限,人们提出了各种自监督分子预训练方法。尽管许多现有方法利用计算机视觉和自然语言处理中常见的预训练任务,但它们经常忽略控制分子的基本物理原理。相比之下,在预训练中应用去噪可以解释为等效的力学习,但有限的噪声分布会给分子分布带来偏差。为了解决这个问题,我们引入了一种称为分数去噪的分子预训练框架,它将噪声设计与力学习等效性施加的约束分离。通过这种方式,噪声变得可定制,允许结合化学先验来显着改进分子分布建模。实验表明,我们的框架始终优于现有方法,在力预测、量子化学性质和结合亲和力任务方面建立了最先进的结果。精细的噪声设计提高了力的准确性和采样覆盖范围,这有助于创建物理一致的分子表示,最终实现卓越的预测性能。
































 京公网安备 11010802027423号
京公网安备 11010802027423号