Nature Microbiology ( IF 20.5 ) Pub Date : 2024-09-18 , DOI: 10.1038/s41564-024-01791-x
Ian R Humphreys 1, 2 , Jing Zhang 3, 4, 5 , Minkyung Baek 6 , Yaxi Wang 7 , Aditya Krishnakumar 1, 2 , Jimin Pei 3, 4, 5 , Ivan Anishchenko 1, 2 , Catherine A Tower 7 , Blake A Jackson 7 , Thulasi Warrier 8, 9, 10 , Deborah T Hung 8, 9, 10 , S Brook Peterson 7 , Joseph D Mougous 7, 11, 12 , Qian Cong 3, 4, 5 , David Baker 1, 2, 11
|
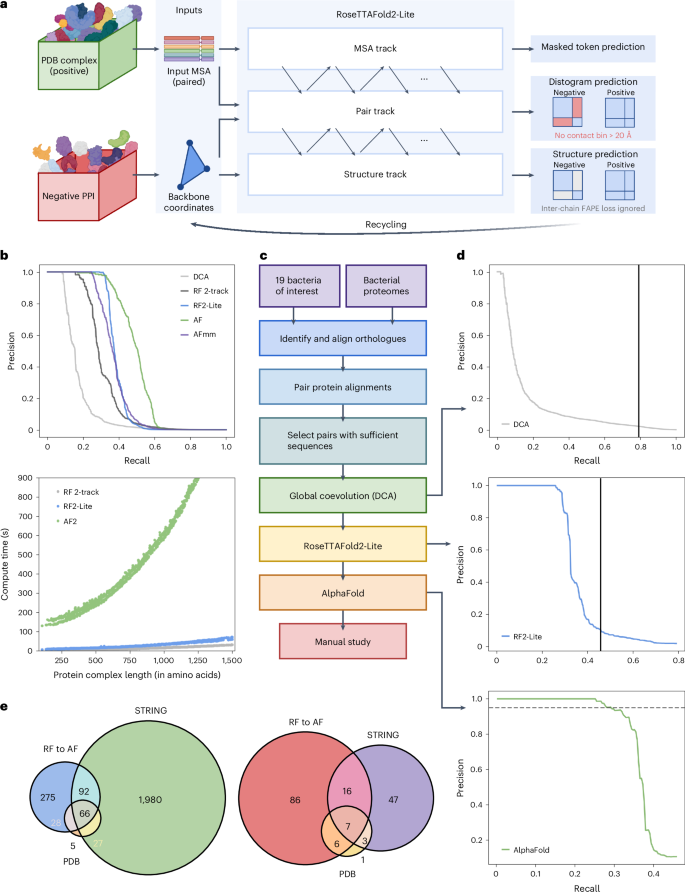
Identification of bacterial protein–protein interactions and predicting the structures of these complexes could aid in the understanding of pathogenicity mechanisms and developing treatments for infectious diseases. Here we developed RoseTTAFold2-Lite, a rapid deep learning model that leverages residue–residue coevolution and protein structure prediction to systematically identify and structurally characterize protein–protein interactions at the proteome-wide scale. Using this pipeline, we searched through 78 million pairs of proteins across 19 human bacterial pathogens and identified 1,923 confidently predicted complexes involving essential genes and 256 involving virulence factors. Many of these complexes were not previously known; we experimentally tested 12 such predictions, and half of them were validated. The predicted interactions span core metabolic and virulence pathways ranging from post-transcriptional modification to acid neutralization to outer-membrane machinery and should contribute to our understanding of the biology of these important pathogens and the design of drugs to combat them.
中文翻译:
通过深度学习揭示人类病原体中的蛋白质相互作用
鉴定细菌蛋白质-蛋白质相互作用并预测这些复合物的结构可能有助于了解致病机制和开发传染病的治疗方法。在这里,我们开发了 RoseTTAFold2-Lite,这是一种快速深度学习模型,它利用残基-残基共进化和蛋白质结构预测来系统地识别和表征蛋白质组范围内的蛋白质-蛋白质相互作用。使用该管道,我们搜索了 19 种人类细菌病原体中的 7800 万对蛋白质,并确定了 1,923 个涉及必需基因的可靠预测复合物和 256 个涉及毒力因子的复合物。其中许多复合体以前并不为人所知;我们实验测试了 12 个这样的预测,其中一半得到了验证。预测的相互作用涵盖核心代谢和毒力途径,从转录后修饰到酸中和再到外膜机制,应该有助于我们了解这些重要病原体的生物学特性和对抗它们的药物设计。

































 京公网安备 11010802027423号
京公网安备 11010802027423号