当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Combined Physics- and Machine-Learning-Based Method to Identify Druggable Binding Sites Using SILCS-Hotspots
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2024-09-16 , DOI: 10.1021/acs.jcim.4c01189 Erik B. Nordquist, Mingtian Zhao, Anmol Kumar, Alexander D. MacKerell, Jr.
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2024-09-16 , DOI: 10.1021/acs.jcim.4c01189 Erik B. Nordquist, Mingtian Zhao, Anmol Kumar, Alexander D. MacKerell, Jr.
|
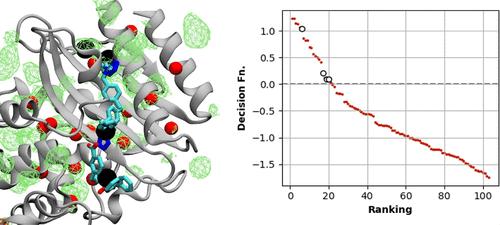
Identifying druggable binding sites on proteins is an important and challenging problem, particularly for cryptic, allosteric binding sites that may not be obvious from X-ray, cryo-EM, or predicted structures. The Site-Identification by Ligand Competitive Saturation (SILCS) method accounts for the flexibility of the target protein using all-atom molecular simulations that include various small molecule solutes in aqueous solution. During the simulations, the combination of protein flexibility and comprehensive sampling of the water and solute spatial distributions can identify buried binding pockets absent in experimentally determined structures. Previously, we reported a method for leveraging the information in the SILCS sampling to identify binding sites (termed Hotspots) of small mono- or bicyclic compounds, a subset of which coincide with known binding sites of drug-like molecules. Here, we build on that physics-based approach and present a ML model for ranking the Hotspots according to the likelihood they can accommodate drug-like molecules (e.g., molecular weight >200 Da). In the independent validation set, which includes various enzymes and receptors, our model recalls 67% and 89% of experimentally validated ligand binding sites in the top 10 and 20 ranked Hotspots, respectively. Furthermore, we show that the model’s output Decision Function is a useful metric to predict binding sites and their potential druggability in new targets. Given the utility the SILCS method for ligand discovery and optimization, the tools presented represent an important advancement in the identification of orthosteric and allosteric binding sites and the discovery of drug-like molecules targeting those sites.
中文翻译:

结合基于物理和机器学习的方法,使用 SILCS 热点识别可成药结合位点
鉴定蛋白质上的可成药结合位点是一个重要且具有挑战性的问题,特别是对于从 X 射线、冷冻电镜或预测结构中可能不明显的隐蔽变构结合位点。通过配体竞争饱和度进行位点鉴定 (SILCS) 方法使用全原子分子模拟(包括水溶液中的各种小分子溶质)来解释靶蛋白的灵活性。在模拟过程中,蛋白质灵活性与水和溶质空间分布的全面采样相结合,可以识别实验确定的结构中不存在的埋藏结合袋。以前,我们报道了一种利用 SILCS 采样中的信息来识别小单环或双环化合物的结合位点(称为热点)的方法,其中的一个子集与药物样分子的已知结合位点一致。在这里,我们建立在这种基于物理的方法之上,并提出了一个 ML 模型,用于根据热点能够容纳药物样分子的可能性(例如,分子量 >200 Da)对热点进行排序。在包括各种酶和受体的独立验证集中,我们的模型分别在排名前 10 和 20 的热点中召回了 67% 和 89% 的实验验证配体结合位点。此外,我们表明该模型的输出决策函数是预测结合位点及其在新靶点中的潜在成药性的有用指标。鉴于 SILCS 方法在配体发现和优化方面的实用性,所提出的工具代表了鉴定正构和变构结合位点以及发现靶向这些位点的药物样分子的重要进步。
更新日期:2024-09-16
中文翻译:
结合基于物理和机器学习的方法,使用 SILCS 热点识别可成药结合位点
鉴定蛋白质上的可成药结合位点是一个重要且具有挑战性的问题,特别是对于从 X 射线、冷冻电镜或预测结构中可能不明显的隐蔽变构结合位点。通过配体竞争饱和度进行位点鉴定 (SILCS) 方法使用全原子分子模拟(包括水溶液中的各种小分子溶质)来解释靶蛋白的灵活性。在模拟过程中,蛋白质灵活性与水和溶质空间分布的全面采样相结合,可以识别实验确定的结构中不存在的埋藏结合袋。以前,我们报道了一种利用 SILCS 采样中的信息来识别小单环或双环化合物的结合位点(称为热点)的方法,其中的一个子集与药物样分子的已知结合位点一致。在这里,我们建立在这种基于物理的方法之上,并提出了一个 ML 模型,用于根据热点能够容纳药物样分子的可能性(例如,分子量 >200 Da)对热点进行排序。在包括各种酶和受体的独立验证集中,我们的模型分别在排名前 10 和 20 的热点中召回了 67% 和 89% 的实验验证配体结合位点。此外,我们表明该模型的输出决策函数是预测结合位点及其在新靶点中的潜在成药性的有用指标。鉴于 SILCS 方法在配体发现和优化方面的实用性,所提出的工具代表了鉴定正构和变构结合位点以及发现靶向这些位点的药物样分子的重要进步。






























 京公网安备 11010802027423号
京公网安备 11010802027423号