当前位置:
X-MOL 学术
›
ACS Macro Lett.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Charge Scaling in Classical Force Fields for Lithium Ions in Polymers
ACS Macro Letters ( IF 5.1 ) Pub Date : 2024-09-13 , DOI: 10.1021/acsmacrolett.4c00368 Dongyue Liang 1 , Yuxi Chen 1 , Chuting Deng 1 , Juan J de Pablo 1
ACS Macro Letters ( IF 5.1 ) Pub Date : 2024-09-13 , DOI: 10.1021/acsmacrolett.4c00368 Dongyue Liang 1 , Yuxi Chen 1 , Chuting Deng 1 , Juan J de Pablo 1
Affiliation
|
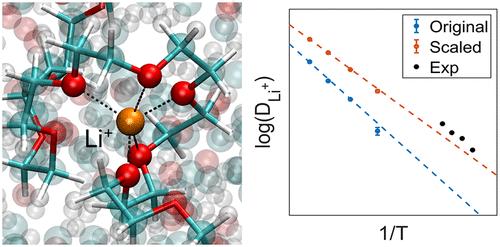
Polymer electrolytes are of interest for applications in energy storage. Molecular simulations of ion transport in polymer electrolytes have been widely used to study the conductivity in these materials. Such simulations have generally relied on classical force fields. A peculiar feature of such force fields has been that in the particular case of lithium ions (Li+), their charge must be scaled down by approximately 20% to achieve agreement with experimental measurements of ion diffusivity. In this work, we present first-principles calculations that serve to justify the charge-scaling factor and van der Waals interaction parameters for Li+ diffusion in poly(ethylene glycol) (PEO) with bistriflimide (TFSI–) counterions. Our results indicate that a scaling factor of 0.79 provides good agreement with DFT calculations over a relatively wide range of Li+ concentrations and temperatures, consistent with past reports where that factor was adjusted by trial and error. We also show that such a scaling factor leads to diffusivities that are in quantitative agreement with experimental measurements.
中文翻译:

聚合物中锂离子的经典力场中的电荷缩放
聚合物电解质在储能中的应用备受关注。聚合物电解质中离子传输的分子模拟已被广泛用于研究这些材料的电导率。此类模拟通常依赖于经典的力场。这种力场的一个独特特征是,在锂离子 (Li+) 的特殊情况下,它们的电荷必须按比例缩小约 20%,才能与离子扩散率的实验测量一致。在这项工作中,我们提出了第一性原理计算,用于证明聚乙二醇 (PEO) 与双立氟胺 (TFSI–) 对离子中 Li+ 扩散的电荷缩放因子和范德华相互作用参数的合理性。我们的结果表明,在相对较宽的 Li+ 浓度和温度范围内,0.79 的比例因子与 DFT 计算具有良好的一致性,这与过去的报告一致,其中该因子是通过反复试验进行调整的。我们还表明,这样的比例因子导致扩散率与实验测量在定量上一致。
更新日期:2024-09-13
中文翻译:
聚合物中锂离子的经典力场中的电荷缩放
聚合物电解质在储能中的应用备受关注。聚合物电解质中离子传输的分子模拟已被广泛用于研究这些材料的电导率。此类模拟通常依赖于经典的力场。这种力场的一个独特特征是,在锂离子 (Li+) 的特殊情况下,它们的电荷必须按比例缩小约 20%,才能与离子扩散率的实验测量一致。在这项工作中,我们提出了第一性原理计算,用于证明聚乙二醇 (PEO) 与双立氟胺 (TFSI–) 对离子中 Li+ 扩散的电荷缩放因子和范德华相互作用参数的合理性。我们的结果表明,在相对较宽的 Li+ 浓度和温度范围内,0.79 的比例因子与 DFT 计算具有良好的一致性,这与过去的报告一致,其中该因子是通过反复试验进行调整的。我们还表明,这样的比例因子导致扩散率与实验测量在定量上一致。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号